Entropy, or Information, Unifies Ecology and Evolution and Beyond

Abstract
:1. A Shared Basis for Ecology and Evolution
- Innovation (e.g., mutation, recombination, divergence and speciation, behavioral innovation)
- Transmission and replication (e.g., inheritance)
- Movement (e.g., migration, pollen dispersal, etc.)
- Adaptation (e.g., selection, behavioral avoidance of harm)
vs.
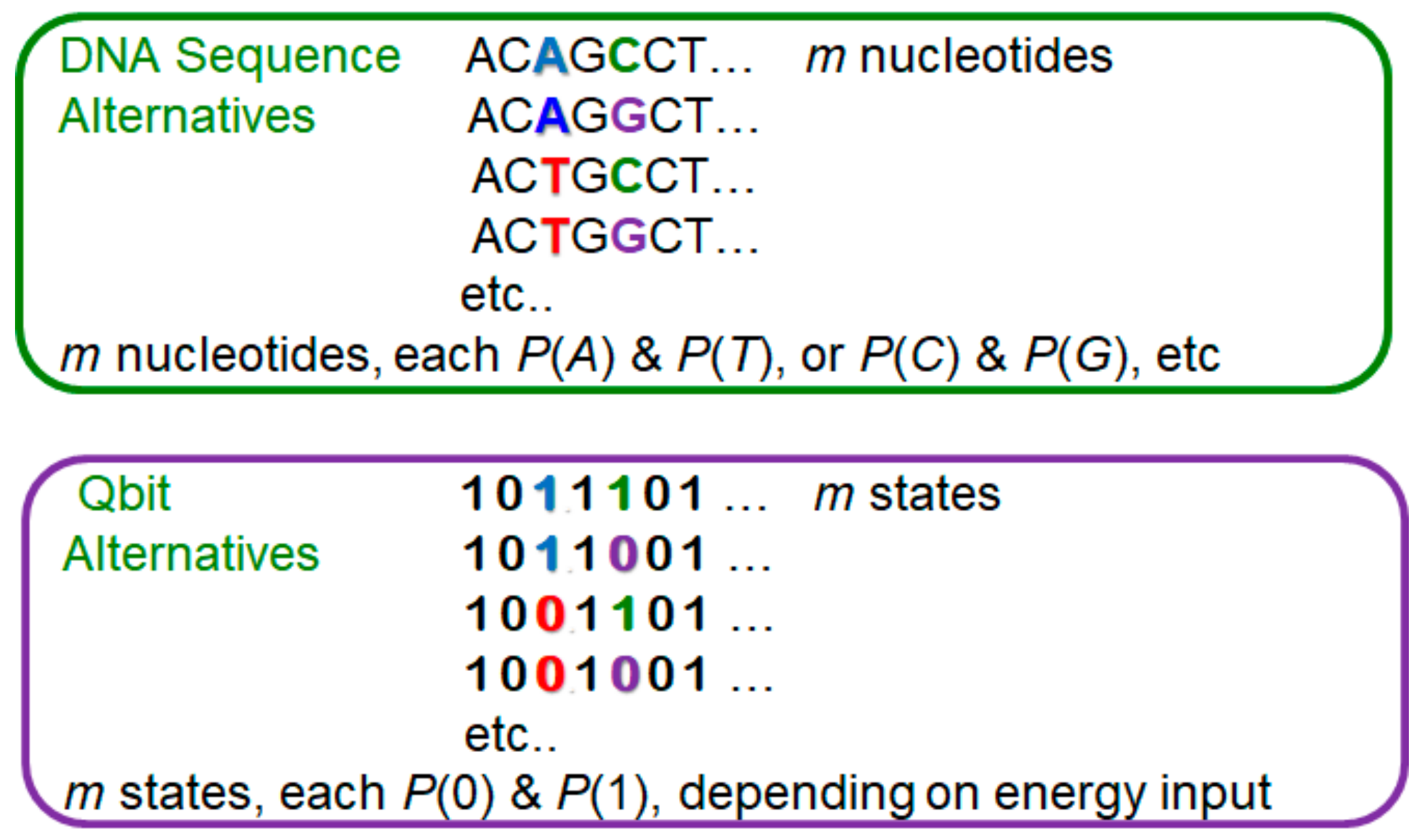
…ACTGCCT…
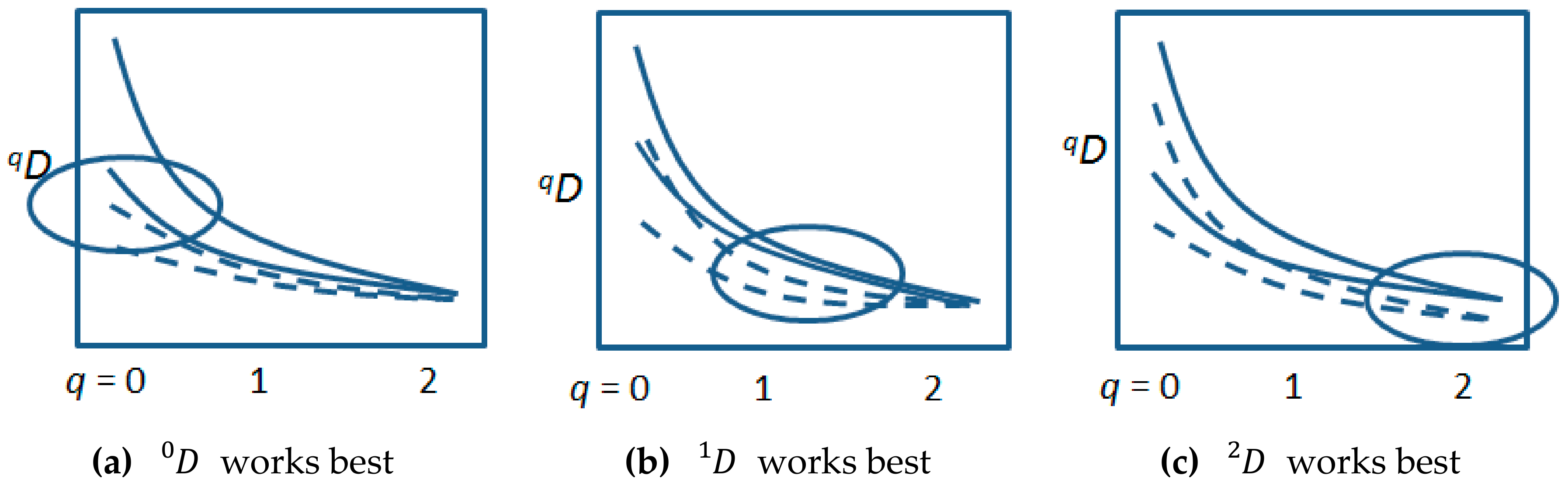
2. Background: Measuring Biological Entropy, Information and Diversity
3. Forecasting Biological Entropy, Information, and Diversity, Based on the Four Processes Common to Ecology and Evolution
 1− 0 1+ 2+, etc. Nowadays this model is also used to approximate innovation in repetitive DNA regions (e.g., ‘microsatellite’ fingerprint DNA). Each model—SNP, IAM, and SMM—is only an approximation, and there are other innovation processes such as insertion or deletion of nucleotides, rearrangements, and epigenetic modification.
1− 0 1+ 2+, etc. Nowadays this model is also used to approximate innovation in repetitive DNA regions (e.g., ‘microsatellite’ fingerprint DNA). Each model—SNP, IAM, and SMM—is only an approximation, and there are other innovation processes such as insertion or deletion of nucleotides, rearrangements, and epigenetic modification.and → and
…ACTGCGT… …ACTGCCT…
then…‘survival juvenile to breeder (e.g., 0.6 survival)’
4. Beyond Ecology and Evolution
- The simple type seen with cells within individuals, or individuals within a population or ecological assemblage, having an exponential rate equation,
- the autocatalytic type seen with some macromolecules, having a hyperbolic rate equation and,
- the template-dependent type, as seen with nucleic acids, having a parabolic rate equation.
5. Extended Ecology and Evolution
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Vellend, M. The Theory of Ecological Communities (MPB-57); Princeton University Press: Princeton, NJ, USA, 2016. [Google Scholar]
- Danchin, E. Avatars of information: Towards an inclusive evolutionary synthesis. Trends Ecol. Evol. 2013, 28, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Frère, C.H.; Krützen, M.; Mann, J.; Connor, R.C.; Bejder, L.; Sherwin, W.B. Social and genetic interactions drive fitness variation in a free-living dolphin population. Proc. Natl. Acad. Sci. USA 2010, 107, 19949–19954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.-H.; Chao, A. Distance-based functional diversity measures and their decomposition: A framework based on Hill numbers. PLoS ONE 2014, 9, e100014. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, W.B.; Chao, A.; Jost, L.; Smouse, P.E. Information Theory Broadens the Spectrum of Molecular Ecology and Evolution. Trends Ecol. Evol. 2017, 32, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Hill, M.O. Diversityand evenness: A unifying notation and its consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef]
- Egizi, A.; Fonseca, D.M. Ecological limits can obscure expansion history: Patterns of genetic diversity in a temperate mosquito in Hawaii. Biol. Invasions 2015, 17, 123–132. [Google Scholar] [CrossRef]
- Chao, A.; Wang, Y.T.; Jost, L. Entropy and the species accumulation curve: A novel entropy estimator via discovery rates of new species. Methods Ecol. Evol. 2013, 4, 1091–1100. [Google Scholar] [CrossRef]
- Chao, A.; Jost, L. Estimating diversity and entropy profiles via discovery rates of new species. Methods Ecol. Evol. 2015, 6, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Buddle, C.M.; Beguin, J.; Bolduc, E.; Mercardo, A.; Sackett, T.E.; Selby, R.D.; Varady-Szabo, H.; Zeran, R.M. The importance and use of taxon sampling curves for comparative biodiversity research with forest arthropod assemblages. Can. Èntomol. 2004, 137, 120–127. [Google Scholar] [CrossRef]
- Day, T. Information entropy as a measure of genetic diversity and evolvability in colonization. Mol. Ecol. 2015, 24, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.A. Universal expressions of population change by the Price equation: Natural selection, information, and maximum entropy production. Ecol. Evol. 2017, 7, 3381–3396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Chen, Y.; Kiralis, J.W.; Collins, R.L.; Wejse, C.; Sirugo, G.; Williams, S.M.; Moore, J.H. An information-gain approach to detecting three-way epistatic interactions in genetic association studies. J. Am. Med. Inform. Assoc. 2013, 20, 630–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, P.; Zhang, A.; Brazeau, D.; Sucheston, L.; Freudenheim, J.L.; Ambrosone, C.; Ramanathan, M. Information-theoretic metrics for visualizing gene-environment interactions. Am. J. Hum. Genet. 2007, 81, 939–963. [Google Scholar] [CrossRef] [PubMed]
- Chanda, P.; Sucheston, L.; Liu, S.; Zhang, A.; Ramanathan, M. Information-theoretic gene-gene and gene-environment interaction analysis of quantitative traits. BMC Genom. 2009, 10, 509. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, S.P. The Unified Neutral Theory of Biodiversity and Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Moore, J.H.; Hu, T. Epistasis analysis using information theory. Epistasis 2015, 1253, 257–268. [Google Scholar]
- Halliburton, R. Introduction to Population Genetics; Pearson: Upper Saddle River, NJ, USA, 2004. [Google Scholar]
- Nielsen, R.; Slatkin, M. An Introduction to Population Genetics Theory and Applications; Sinauer: Sunderland, MA, USA, 2013. [Google Scholar]
- Meirmans, P.G.; Hedrick, P.W. Assessing population structure: Fst and related measures. Mol. Ecol. Resour. 2011, 11, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Jost, L. Gst and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [PubMed]
- Fisher, R.A.; Corbet, A.S.; Williams, C.B. The relation between the number of species and the number of individuals in a random sample of an animal population. J. Anim. Ecol. 1943, 12, 42–58. [Google Scholar] [CrossRef]
- Preston, F.W. The commonness, and rarity, of species. Ecology 1948, 29, 254–283. [Google Scholar] [CrossRef]
- Ewens, W. Mathematical Population Genetics; Springer-Verlag: Berlin, Germany, 1979. [Google Scholar]
- Leinster, T.; Cobbold, C. Measuring diversity: The importance of species similarity. Ecology 2012, 93, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, A.; Chiu, C.-H.; Jost, L. Unifying species diversity, phylogenetic diversity, functional diversity, and related similarity and differentiation measures through Hill numbers. Annual Rev. Ecol. Evol. Syst. 2014, 45, 297–324. [Google Scholar] [CrossRef]
- Jost, L.; DeVries, P.; Walla, T.; Greeney, H.; Chao, A.; Ricotta, C. Partitioning diversity for conservation analyses. Divers. Distrib. 2010, 16, 65–76. [Google Scholar] [CrossRef]
- Jost, J.; Archer, F.; Flanagan, S.; Gaggiotti, O.; Hoban, S.; Latch, E. Differentiation measures for conservation genetics. Evol. Appl. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Ewens, W.J. The sampling theory of selectively neutral alleles. Theor. Popul. Biol. 1972, 3, 87–112. [Google Scholar] [CrossRef]
- Sherwin, W.B.; Jabot, F.; Rush, R.; Rossetto, M. Measurement of biological information with applications from genes to landscapes. Mol. Ecol. 2006, 15, 2857–2869. [Google Scholar] [CrossRef] [PubMed]
- Dewar, R.C.; Sherwin, W.B.; Thomas, E.; Holleley, C.E.; Nichols, R.A. Predictions of single-nucleotide polymorphism differentiation between two populations in terms of mutual information. Mol. Ecol. 2011, 20, 3156–3166. [Google Scholar] [CrossRef] [PubMed]
- Chao, A.; Jost, L.; Hsieh, T.C.; Ma, K.H.; Sherwin, W.B.; Rollins, L.A. Expected Shannon entropy and Shannon differentiation between subpopulations for neutral genes under the finite island model. PLoS ONE 2015, 10, e0125471. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, G.D.; Jabot, F.; Gunn, M.R.; Sherwin, W.B. Novel uses for equations: Predicting Shannon’s information for genes in finite populations. Conserv. Genet. Resour. 2018. submitted. [Google Scholar]
- Iwasa, Y. Free fitness that always increases in evolution. J. Theor. Biol. 1988, 135, 265–281. [Google Scholar] [CrossRef]
- De Vladar, H.P.; Barton, N.H. The contribution of statistical physics to evolutionary biology. Trends Ecol. Evol. 2011, 26, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwin, W.B. Review: Entropy and information approaches to genetic diversity and its expression: Genomic geography. Entropy 2010, 12, 1765–1798. [Google Scholar] [CrossRef]
- Coyne, J.; Orr, H.A. Speciation; Sinauer: Sunderland, MA, USA, 2004. [Google Scholar]
- Thibert-Plante, X.; Gavrilets, S. Evolution of mate choice and the so-called magic traits in ecological speciation. Ecol. Lett. 2013, 16, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosindell, J.; Cornell, S.J.; Hubbell, S.P.; Etienne, R.S. Protracted speciation revitalizes the neutral theory of biodiversity. Ecol. Lett. 2010, 13, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.H.; Hu, T. Epistasis analysis using information theory. In Epistasis: Methods and Protocols, Methods in Molecular Biology; Moore, J.H., Williams, S.M., Eds.; Springer Science + Business Media: New York, NY, USA, 2015; Volume 1253, pp. 257–268. [Google Scholar]
- Piñero, J.; Solé, R. Nonequilibrium entropic bounds for Darwinian replicators. Entropy 2018, 20, 98. [Google Scholar] [CrossRef]
- Grzywacz, B.; Lehmann, A.W.; Chobanov, D.; Lehmann, G.U.C. Multiple origin of flightlessness in Phaneropterinae bushcrickets and redefinition of the tribus Odonturini (Orthoptera: Tettigonioidea: Phaneropteridae). Org. Divers. Evol. 2018, 1–3. [Google Scholar] [CrossRef]
- Andersen, N.M. The evolution of wing polymorphism in water striders (Gerridae): A phylogenetic approach. Oikos 1993, 67, 433–443. [Google Scholar] [CrossRef]
- Crnokrak, P.; Roff, D.A. The genetic basis of the trade-off between calling and wing morph in males of the cricket Gryllus firmus. Evolution 1998, 52, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- James, M.K.; Armsworth, P.R.; Mason, L.B.; Bode, L. The structure of reef fish metapopulations: Modeling larval dispersal and retention patterns. Proc. R. Soc. Lond. B 2002, 269, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- López-Duarte, P.C.; Carson, H.S.; Cook, G.S.; Fodrie, F.J.; Becker, B.J.; Dibacco, C.; Levin, L.A. What controls connectivity? An empirical, multi-species approach. Integr. Comp. Biol. 2012, 52, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Castorani, M.C.N.; Reed, D.C.; Raimondi, P.T.; Alberto, F.; Bell, T.W.; Cavanaugh, K.C.; Siegel, D.A.; Simons, R.D. Fluctuations in population fecundity drive variation in demographic connectivity and metapopulation dynamics. Proc. R. Soc. B 2017, 284, 20162086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, T.J.; Whittaker, R.J. Neutral theory and the species abundance distribution: Recent developments and prospects for unifying niche and neutral perspectives. Ecol. Evol. 2014, 4, 2263–2277. [Google Scholar] [CrossRef] [PubMed]
- Elith, J.; Phillips, S.J.; Hastie, T.; Dudık, M.; Chee, Y.E.; Yates, C.J. A statistical explanation of MaxEnt for ecologists. Divers. Distrib. 2011, 17, 43–57. [Google Scholar] [CrossRef]
- Bessa, R.J.; Miranda, V.; Gama, J. Entropy and correntropy against minimum square error in offline and online three-day ahead wind power forecasting. IEEE Trans. Power Syst. 2009, 24, 1657–1666. [Google Scholar] [CrossRef]
- Skene, K.R. Life’s a gas: A thermodynamic theory of biological evolution. Entropy 2015, 17, 5522–5548. [Google Scholar] [CrossRef]
- Dewar, R.C.; Porte, A. Statistical mechanics unifies different ecological patterns. J. Theor. Biol. 2008, 251, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borlase, S.C.; Loebel, D.A.; Frankham, R.; Nurthen, R.K.; Briscoe, D.A.; Daggard, G.E. Modeling problems in conservation genetics using captive drosophila populations—Consequences of equalization of family sizes. Conserv. Biol. 1993, 7, 122–131. [Google Scholar] [CrossRef]
- Schlötterer, C.; Kofler, R.; Versace, E.; Tobler, T.; Franssen, S.U. Combining experimental evolution with next-generation sequencing: A powerful tool to study adaptation from standing genetic variation. Heredity 2015, 114, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Vandepitte, K.; De Meyer, T.; Helsen, K.; Van Acker, K.; Roldán-Ruiz, I.; Mergeay, J.; Honnay, O. Rapid genetic adaptation precedes the spread of an exotic plant species. Mol. Ecol. 2014, 23, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.; Nichols, R.A. Evaluating loci for use in the geentic analysis of popualtion structure. Proc. R. Soc. Lond. 1996, 263, 1619–1626. [Google Scholar] [CrossRef]
- Chao, A.; Chiu, C.-H.; Villéger, S.; Sun, I.-F.; Thorn, S.; Lin, Y.; Chiang, J.-M.; Sherwin, W.B. An attribute-diversity approach to functional diversity, functional beta diversity, and related (dis)similarity measures. Ecol. Monogr. 2018. submitted. [Google Scholar]
- Dodig-Crnkovica, G. Nature as a network of morphological infocomputational processes for cognitive agents. Eur. Phys. J. Spec. Top. 2017, 226, 181. [Google Scholar] [CrossRef]
- Barton, N.H.; De Vladar, H.P. Statistical mechanics and the evolution of polygenic quantitative traits. Genetics 2009, 181, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Popa, R.; Cimpoiasu, V.M. Prebiotic Competition between Information Variants, With Low Error Catastrophe Risks. Entropy 2015, 17, 5274–5287. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.X. Quantum-inspired evolutionary algorithms: A survey and empirical study. J. Heuristics 2011, 17, 303–351. [Google Scholar] [CrossRef]
- Hamblin, S. On the practical usage of genetic algorithms in ecology and evolution. Methods Ecol. Evol. 2013, 4, 184–194. [Google Scholar] [CrossRef]
- Miller, S.L.; Urey, H.C. Organic Compound Synthesis on the Primitive Earth. Science 1959, 130, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Yukalov, V.I.; Sornette, D. Quantum probability and quantum decision-making. Phil. Trans. R. Soc. A 2016, 374, 20150100. [Google Scholar] [CrossRef] [PubMed]
- Paixão, T.; Heredia, J.P.; Sudholt, D.; Trubenová, B. Towards a runtime comparison of natural and artificial evolution. Algorithmica 2017, 78, 681. [Google Scholar] [CrossRef]
- Zhao, L.; Charlesworth, B. Resolving the conflict between associative overdominance and background selection. Genetics 2016, 203, 1315–1334. [Google Scholar] [CrossRef] [PubMed]
- Blum, C.; Roli, A. Metaheuristics in combinatorial optimization. Comput. Surv. 2003, 35, 268–308. [Google Scholar] [CrossRef]
- Von Kodolitsch, Y.; Berger, J.; Rogan, P.K. Predicting severity of haemophilia A and B splicing mutations by information analysis. Haemophilia 2006, 12, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A. Quantum Computing for beginners. Proc Congr. Evol. Comput. 1999, 1999, 2231–2238. [Google Scholar]
- Robertson, A. Selection for heterozygotes in small populations. Genetics 1962, 47, 1291–1300. [Google Scholar] [PubMed]
- Sutton, J.T.; Nakagawa, S.; Robertson, B.C.; Jamieson, I.G. Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol. Ecol. 2011, 20, 4408–4420. [Google Scholar] [CrossRef] [PubMed]
- Lewin, H.A.; Robinson, G.E.; Kress, W.J.; Baker, W.J.; Coddington, J.; Crandall, K.A.; Durbin, R.; Edwards, S.V.; Forest, F.; Gilbert, M.T.P.; et al. Earth BioGenome Project: Sequencing life for the future of life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Entropy Effective Number | ECOLOGY: Variant Species in an Assemblage | EVOLUTION: Variant Molecules (Genes) within Species |
|---|---|---|
| (a) Measurement | ||
| = Count of types − 1 = Count of types | Used, but has very wide confidence limits, even with modern corrections [9,10]. | |
Where p values are the proportions of the different variants | The most common frequency-sensitive measure [11]. | Rarely used until recently [5]. Related measures are proposed as a primary measure of evolvability [12,13]. Commonly used for analyzing networks of physically linked or functionally interacting genes [5,14,15,16,17]. |
| Some use [18] | The most common measure (Heterozygosity, Nucleotide diversity, STRUCTURE, AMOVA, FST, G”ST, DEST, etc.) [19,20,21,22,23,24]. | |
| (b) Forecasts from Underlying Processes | ||
| = Count of types − 1 = Count of types | No forecasts from underlying processes; some from curve-fitting [25,26]. | Some forecasts, with underlying transmission and innovation only [27]. |
| Forecasts are available to be transferred from Molecular Ecology [5]. | Forecasting ability now close to matching that for q = 2 [5]. Further details are in Table 2. | |
| Some forecasts transferred from Molecular Ecology, but only with underlying transmission and innovation, no adaptation [18]. | Extensive ability to forecast under a wide range of conditions for all underlying processes: Innovation, Transmission, Movement, and Adaptation. Forecasts are often based on gas diffusion theory, e.g., Fokker–Planck Equation (see summaries in textbooks [19,20]) | |
| UNDERLYING PROCESSES | Space and Time Scales | |||
|---|---|---|---|---|
| α Within-Locality | β between-Locality | |||
| Finite Size, at Equilibrium | Dynamic: Non-Equilibrium | Finite Size, at Equilibrium | Dynamic: Non-Equilibrium | |
| INNOVATION | Innovation mechanisms—SNP, IAM and SMM—are defined and described further in the text, including the relationships between forecasts for molecules within one species (described in this table) and forecasts for species in assemblages | |||
| TRANSMISSION Neutral variants (i.e., no effect on adaptation) with stochasticity | SNP [34] IAM [33,35] SMM [33,35] | SNP, IAM, SMM [36] SNP [34] | SNP [34] IAM [33,35] SMM [33,35] | SNP [34] |
| MOVEMENT Neutral variants, with dispersal between locations | - | - | SNP [34] IAM [33,35] SMM [33,35] | SNP [34] |
| ADAPTATION Continuous heritable variants, e.g., reproductive rate or gene expression patterns | [5,37,38] | [5,37,38] | Not Yet | Not Yet |
| ADAPTATION Discrete heritable variants, e.g., DNA alleles or haplotypes | ‘Balancing’ selection that maintains more than one variant [39] | ‘Directional’ selection that favors a single variant ([12,13] and Supp. S4, S5 of review [5]) | Not Yet | Not Yet |
| System | Common Processes for Information | |||
|---|---|---|---|---|
| Innovation | Transmission | Adaptation | Movement | |
| Prebiotic (may be continuing slowly in current physical environment) | Many years? [65] | Seconds, or longer, rate depends upon type of interactions [43] | Speed would depend upon relative rates of innovation and competitive interactions [62]. | Probably occurs, at least involuntarily in currents, etc. |
| Biomolecules—acting individually | Seconds, or longer | Seconds, or longer, rate depends upon type of interactions [43] | Seconds, or longer | Seconds, or longer |
| Biomolecules—as basis of biological evolution | Generations [19,20] | Generations [19,20] | Generations [19,20] | Generations [19,20] |
| Neural networks and Behavioral responses driven by neurons | Seconds | Seconds | Seconds (or longer with a group of individuals [66] | Seconds, or longer |
| Species | Usually 1000’s of generations [1,18,40] | Usually 1000’s of generations [1,18,40] | Usually 1000’s of generations [1,40] | Usually 1000’s of generations [1,18,40] |
| Algorithms and machines | Seconds to Hours [63,64,67] | Seconds to Hours [63,64,67] | Seconds to Hours [63,64,67] | Seconds to Hours e.g., Self-driving cars, Mars rovers, Computer viruses |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherwin, W.B. Entropy, or Information, Unifies Ecology and Evolution and Beyond. Entropy 2018, 20, 727. https://doi.org/10.3390/e20100727
Sherwin WB. Entropy, or Information, Unifies Ecology and Evolution and Beyond. Entropy. 2018; 20(10):727. https://doi.org/10.3390/e20100727
Chicago/Turabian StyleSherwin, William Bruce. 2018. "Entropy, or Information, Unifies Ecology and Evolution and Beyond" Entropy 20, no. 10: 727. https://doi.org/10.3390/e20100727
APA StyleSherwin, W. B. (2018). Entropy, or Information, Unifies Ecology and Evolution and Beyond. Entropy, 20(10), 727. https://doi.org/10.3390/e20100727