A Chemo-Mechanical Model of Diffusion in Reactive Systems


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanical Deformation and Diffusion
2.1. Kinematics of Deformation
2.2. Mass Balance
2.3. Helmholtz Free-Energy Density of the System
2.3.1. Elastic Energy Contribution
2.3.2. Configurational Energy Contribution
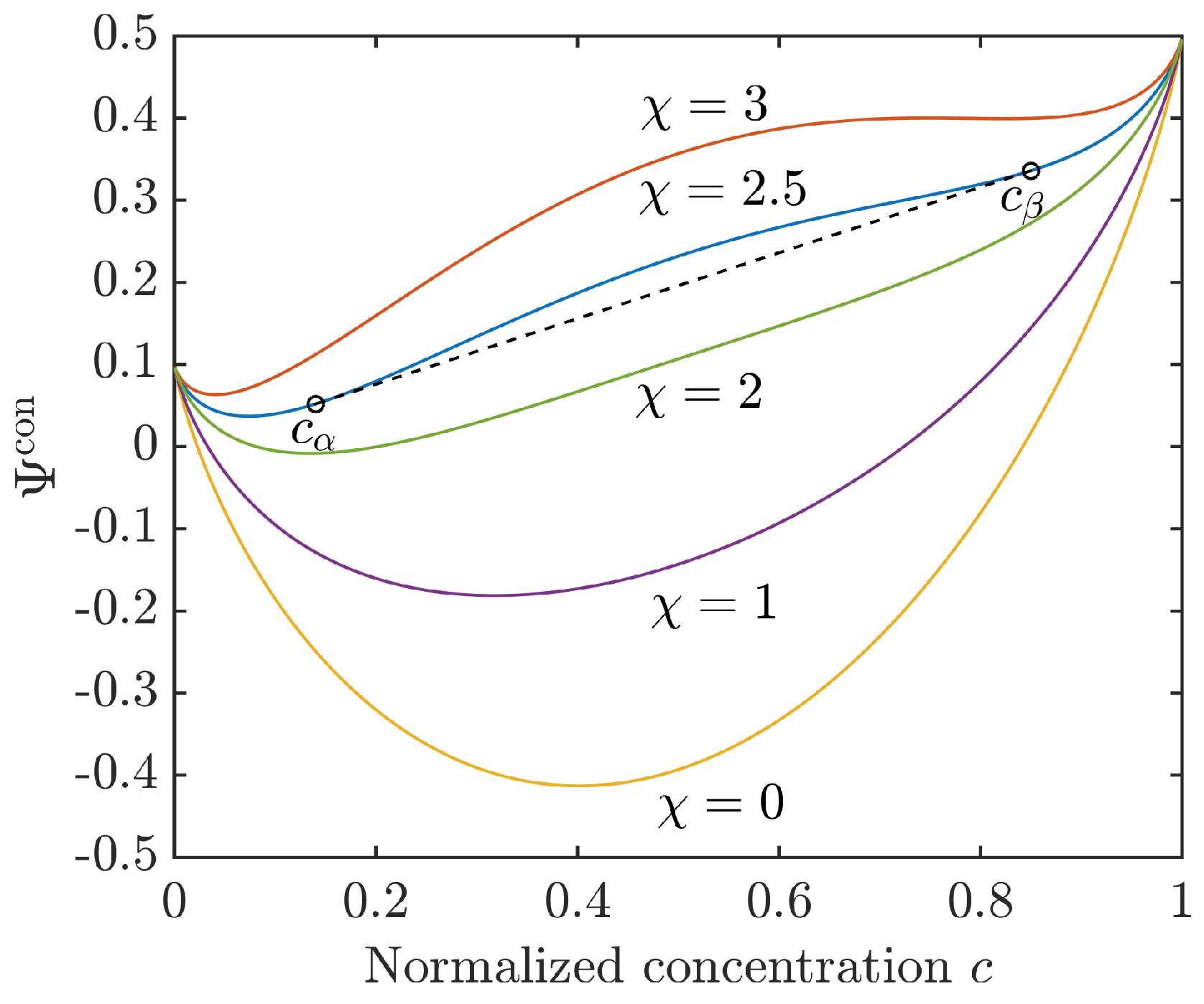
2.3.3. Interfacial Energy Contribution
2.4. Mechanical Stresses and Chemical Potential
3. Chemical Reactions
3.1. Kinetics of Chemical Reactions
3.2. Thermodynamical Model for Reaction-Diffusion Systems
3.3. Formulation of the Problem
4. Computational Studies of Elastic Multicomponent Reaction-Diffusion Systems
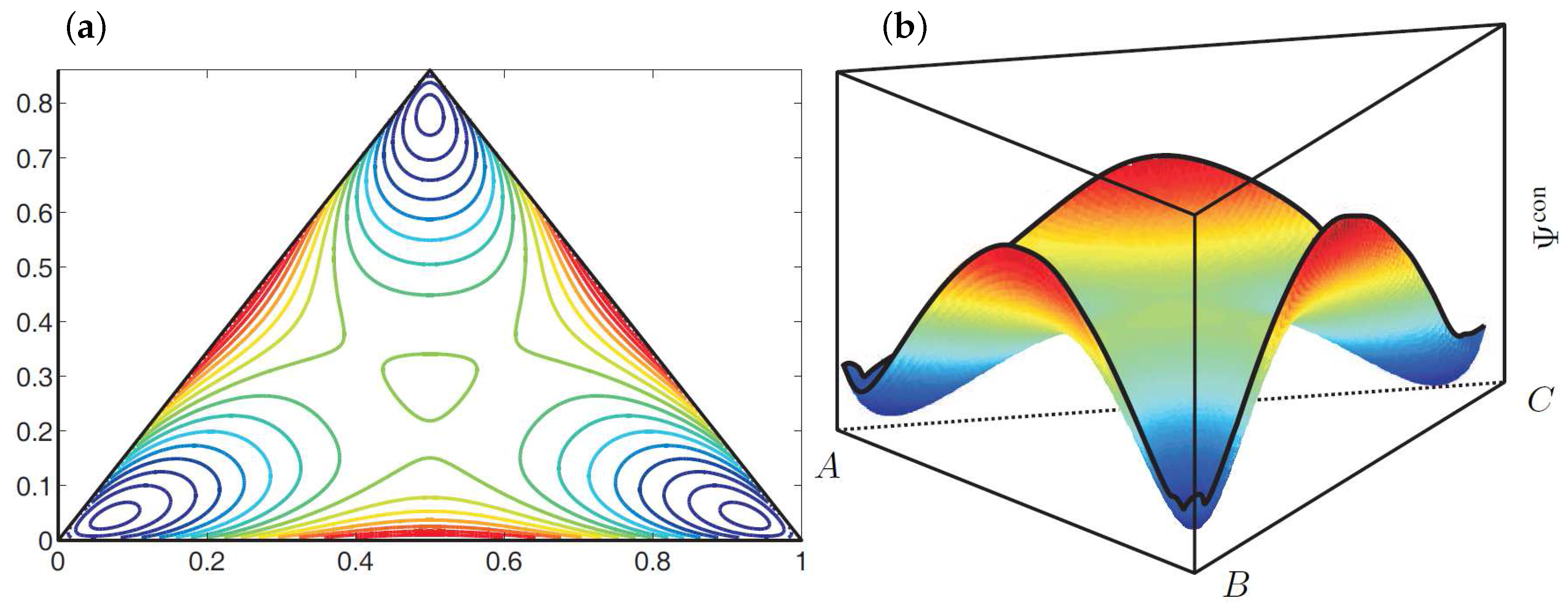
4.1. Evolution of a Ternary Reaction-Diffusion System
4.1.1. Modelling the System
4.1.2. Choice of Parameters
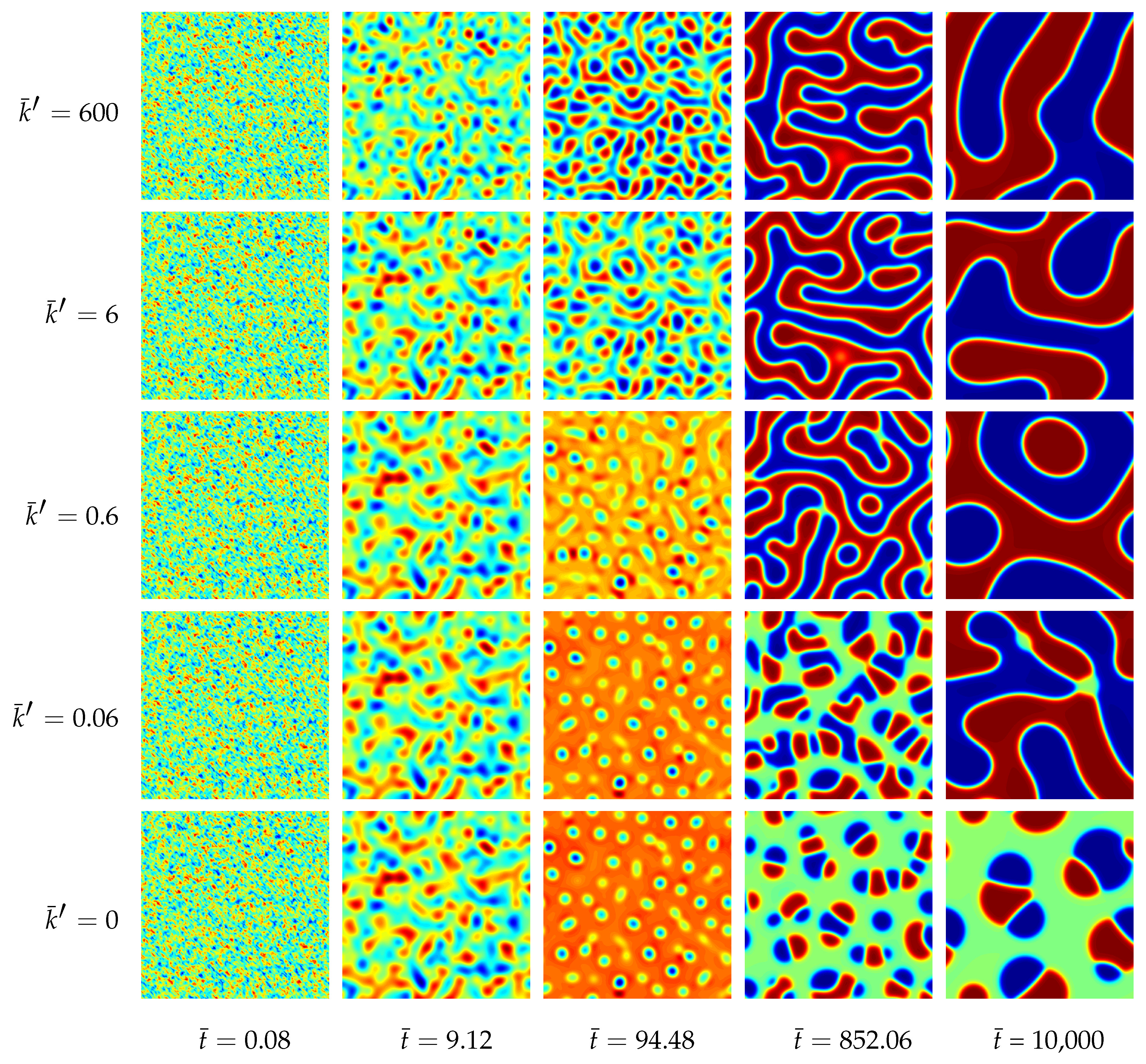
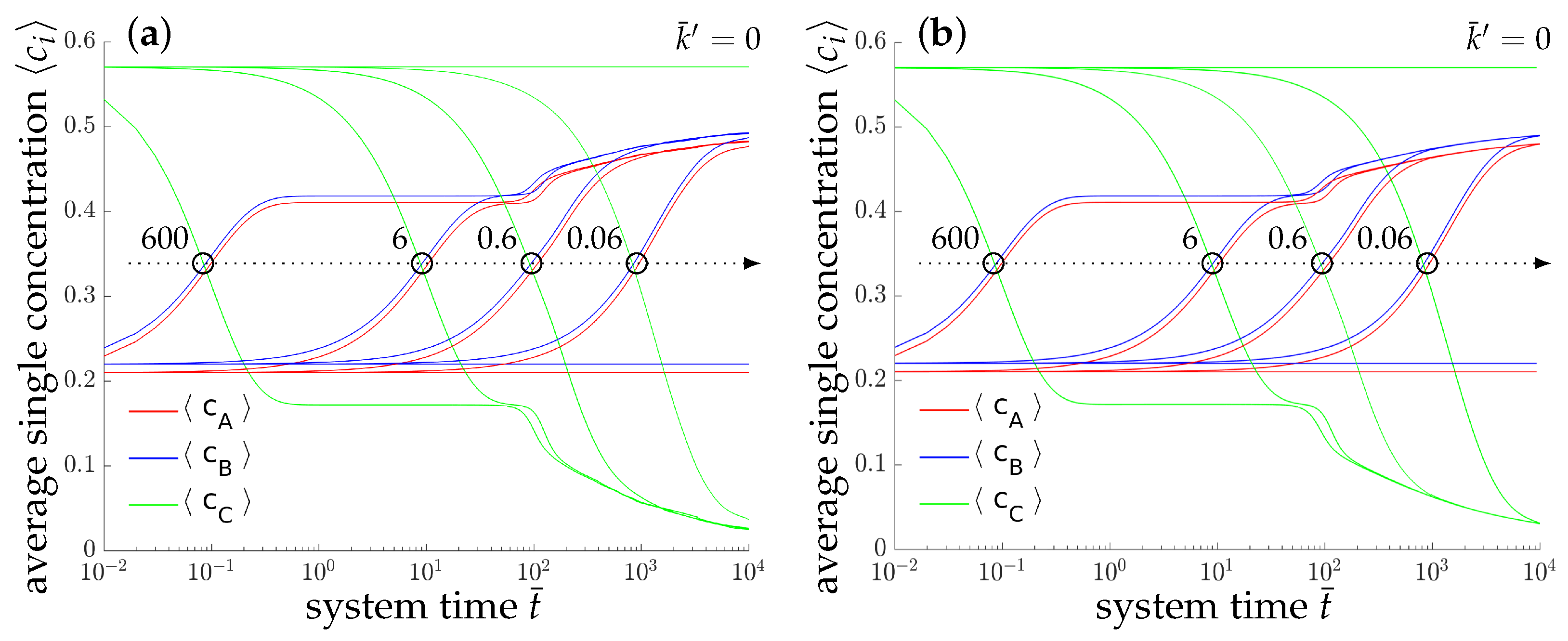
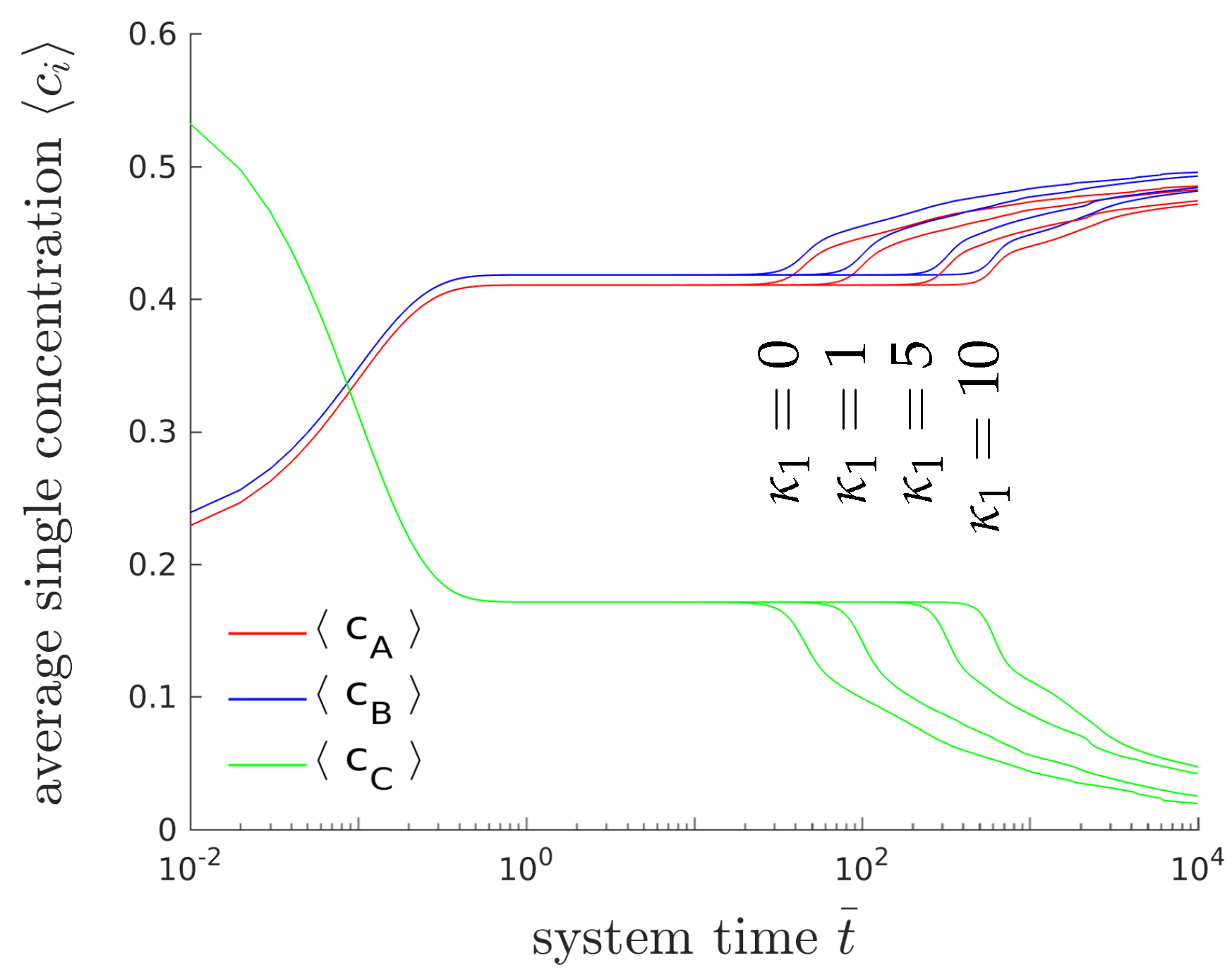
4.1.3. Results of the Two-Dimensional Simulations
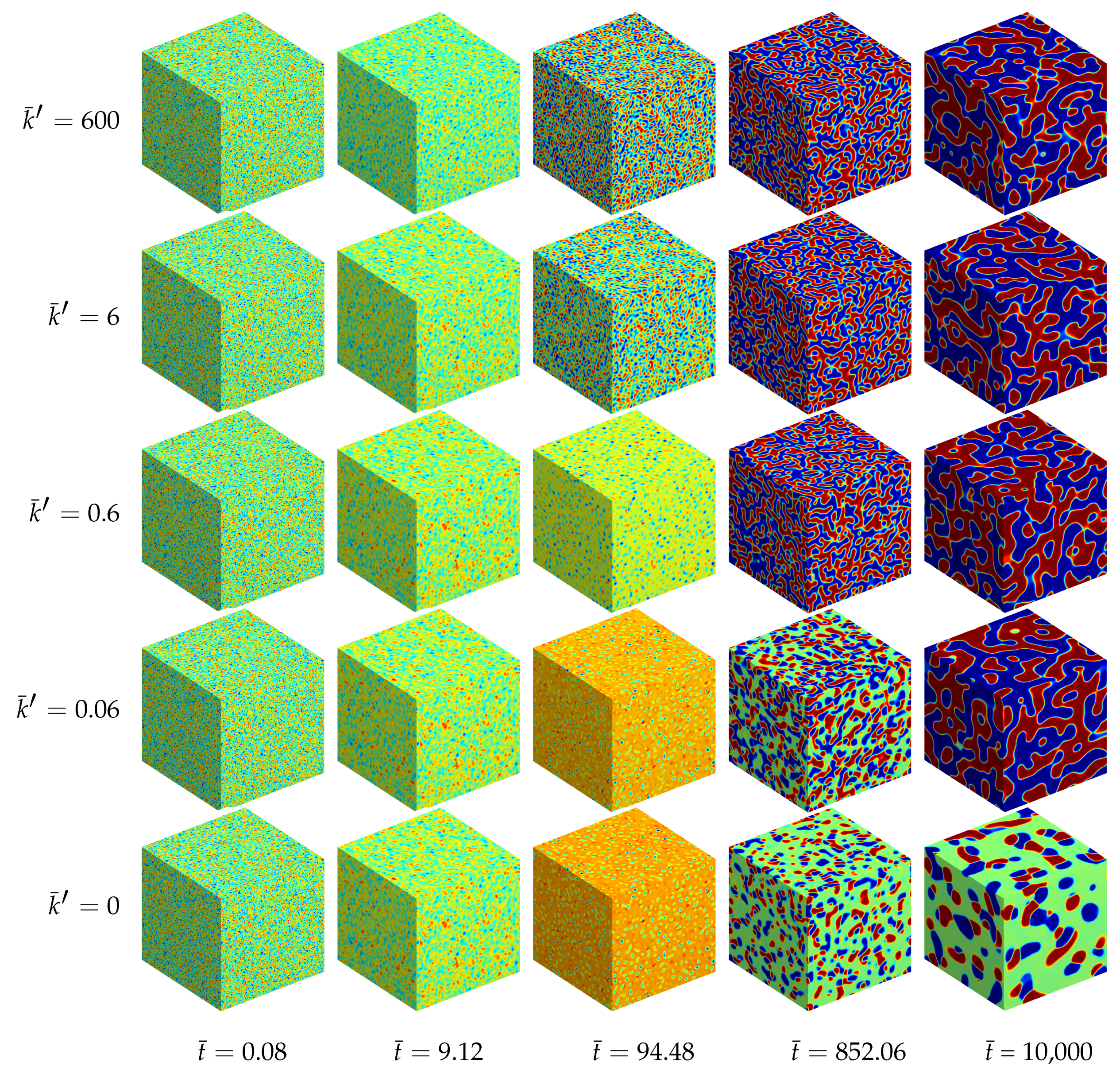
4.1.4. Results of the Three-Dimensional Simulations
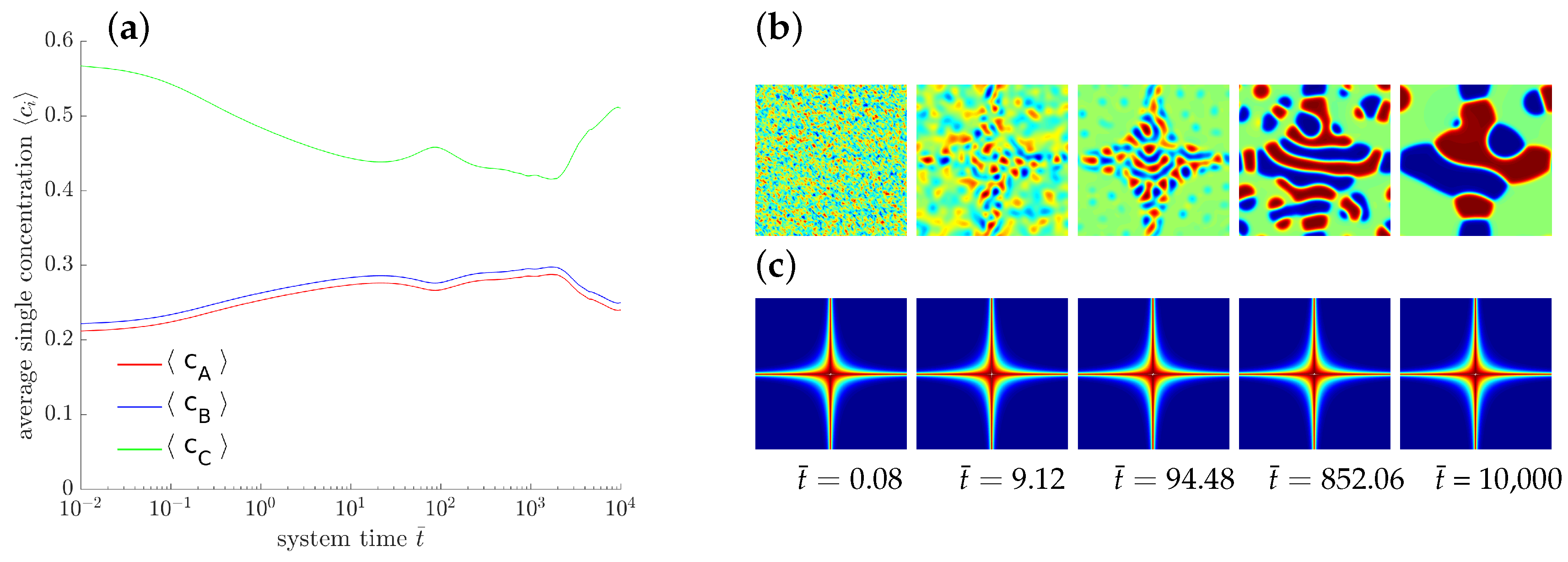
4.1.5. Parametric Studies
4.2. Evolution of Elastic Phase-Separating Systems
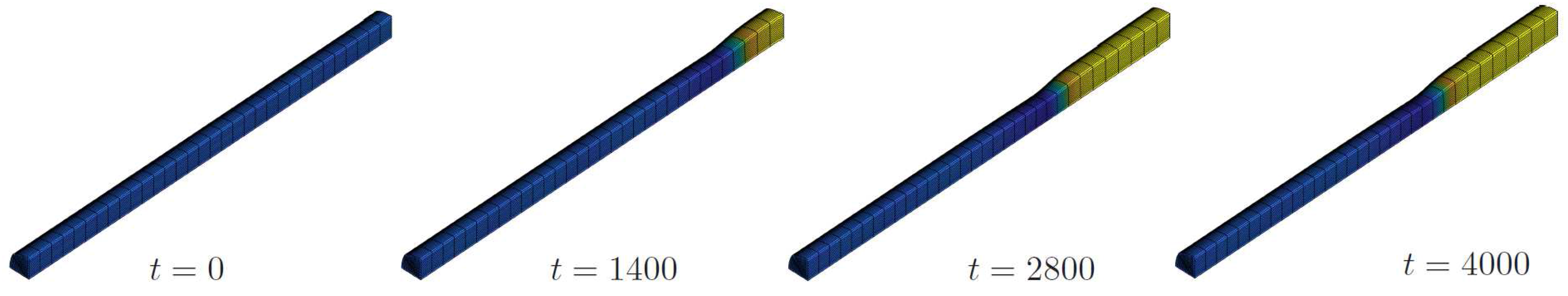
4.2.1. A Rod under to Incoming Flux
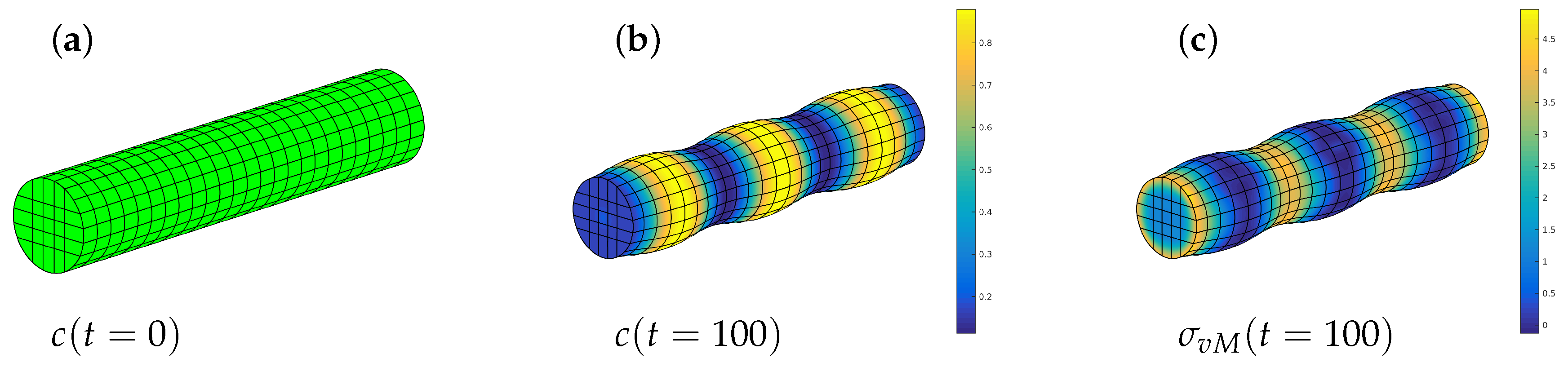
4.2.2. Phase Decomposition in an Elastic Rod
5. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tikhomirov, V.M. A Study of the Diffusion Equation with Increase in the Amount of Substance, and its Application to a Biological Problem. In Selected Works of A. N. Kolmogorov: Volume I: Mathematics and Mechanics; Tikhomirov, V.M., Ed.; Mathematics and Its Applications (Soviet Series); Springer: Dordrecht, The Netherlands, 1991; pp. 242–270. [Google Scholar]
- Fisher, R.A. The wave of advance of advantageous genes. Ann. Hum. Genet. 1937, 7, 355–369. [Google Scholar] [CrossRef]
- Cencini, M.; Lopez, C.; Vergni, D. Reaction-Diffusion Systems: Front Propagation and Spatial Structures. In The Kolmogorov Legacy in Physics; Livi, R., Vulpiani, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; Volume 636, pp. 187–210. [Google Scholar]
- Takagi, H.; Kaneko, K. Pattern dynamics of a multi-component reaction diffusion system: Differentiation of replicating spots. Int. J. Bifurc. Chaos 2002, 12, 2579–2598. [Google Scholar] [CrossRef]
- Mendez, V.; Fedotov, S.; Horsthemke, W. Reaction-Transport Systems. In Mesoscopic Foundations, Fronts, and Spatial Instabilities, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 3–419. [Google Scholar]
- De Groot, S.; Mazur, P. Non-Equilibrium Thermodynamics; Courier Corporation: Amsterdam, The Netherlands, 1962; pp. 197–234. [Google Scholar]
- Larchté, F.C.; Cahn, J.W. The effect of self-stress on diffusion in solids. Acta Metall. 1982, 30, 1835–1845. [Google Scholar] [CrossRef]
- Eckart, C. The thermodynamics of irreversible processes. IV. The theory of elasticity and anelasticity. Phys. Rev. 1948, 73, 373–380. [Google Scholar] [CrossRef]
- Beaulieu, L.Y.; Eberman, K.W.; Turner, R.L.; Krause, L.J.; Dahn, J.R. Colossal reversible volume changes in lithium alloys. Electrochem. Solid-State Lett. 2001, 4, 137–140. [Google Scholar] [CrossRef]
- Natsiavasa, P.P.; Weinberg, K.; Rosato, D.; Ortiz, M. Effect of Prestress on the Stability of Electrode-Electrolyte Interfaces during Charging in Lithium Batteries. J. Mech. Phys. Solids 2016, 95, 92–111. [Google Scholar] [CrossRef]
- O’Connell, J.; Haile, J. Thermodynamics: Fundamentals for Applications; Cambridge University Press: New York, NY, USA, 2005; pp. 1–674. [Google Scholar]
- Marsden, J.E.; Ratiu, T.S. Introduction to Mechanics and Symmetry: A Basic Exposition of Classical Mechanical Systems, 2nd ed.; Texts in Applied Mathematics; Springer: New York, NY, USA, 2002; pp. 1–517. [Google Scholar]
- Müller, P. Glossary of terms used in physical organic chemistry. (IUPAC recommendations 1994). Pure Appl. Chem. 1994, 66, 1077–1184. [Google Scholar]
- Cross, M.; Greenside, H. Pattern Formation and Dynamics in Nonequilibrium Systems; Cambridge University Press: Cambridge, UK, 2009; pp. 1–495. [Google Scholar]
- Anders, D.; Weinberg, K. Modeling of multicomponent reactive systems. Tech. Mech. 2012, 32, 105–112. [Google Scholar]
- Anders, D.; Dittmann, M.; Weinberg, K. A higher-order finite element approach to the Kuramoto-Sivashinsky equation. J. Appl. Math. Mech. 2012, 92, 599–607. [Google Scholar] [CrossRef]
- Hesch, C.; Schuß, S.; Dittmann, M.; Franke, M.; Weinberg, K. Isogeometric analysis and hierarchical refinement for higher-order phase-field models. Comput. Methods Appl. Mech. Eng. 2016, 303, 185–207. [Google Scholar] [CrossRef]
- Anders, D.; Weinberg, K. A variational approach to the decomposition of unstable viscous fluids and its consistent numerical approximation. J. Appl. Math. Mech. 2011, 91, 609–629. [Google Scholar] [CrossRef]
- Anders, D.; Weinberg, K. Numerical simulation of diffusion induced phase separation and coarsening in binary alloys. Comput. Mater. Sci. 2011, 50, 1359–1364. [Google Scholar] [CrossRef]
- Glotzer, S.; Di Marzio, E.; Muthukumar, M. Reaction-controlled morphology of phase-separating mixtures. Phys. Rev. Lett. 1995, 74, 2034–2037. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Matchariyakul, N.; Yase, K.; Kitano, T. Morphology development through an interfacial reaction in ternary immiscible polymer blends. Macromolecules 1997, 30, 3664–3670. [Google Scholar] [CrossRef]
- Anders, D.; Weinberg, K. An extended stochastic diffusion model for ternary mixtures. Mech. Mater. 2013, 56, 122–130. [Google Scholar] [CrossRef]
- Zhao, Y.; Stein, P.; Xu, B.-X. Isogeometric analysis of mechanically coupled Cahn-Hilliard phase segregation in hyperelastic electrodes of Li-ion batteries. Comput. Methods Appl. Mech. Eng. 2015, 297, 325–347. [Google Scholar] [CrossRef]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinberg, K.; Werner, M.; Anders, D. A Chemo-Mechanical Model of Diffusion in Reactive Systems. Entropy 2018, 20, 140. https://doi.org/10.3390/e20020140
Weinberg K, Werner M, Anders D. A Chemo-Mechanical Model of Diffusion in Reactive Systems. Entropy. 2018; 20(2):140. https://doi.org/10.3390/e20020140
Chicago/Turabian StyleWeinberg, Kerstin, Marek Werner, and Denis Anders. 2018. "A Chemo-Mechanical Model of Diffusion in Reactive Systems" Entropy 20, no. 2: 140. https://doi.org/10.3390/e20020140
APA StyleWeinberg, K., Werner, M., & Anders, D. (2018). A Chemo-Mechanical Model of Diffusion in Reactive Systems. Entropy, 20(2), 140. https://doi.org/10.3390/e20020140