Solving Stochastic Reaction Networks with Maximum Entropy Lagrange Multipliers

Abstract
:1. Introduction
2. Materials and Methods/Theory
2.1. Lagrange Multiplier Equations
2.1.1. Connect Moments to Lagrange Multipliers
2.1.2. Time Derivatives
2.2. Initial Value from Moments to Lagrange Multipliers
3. Results
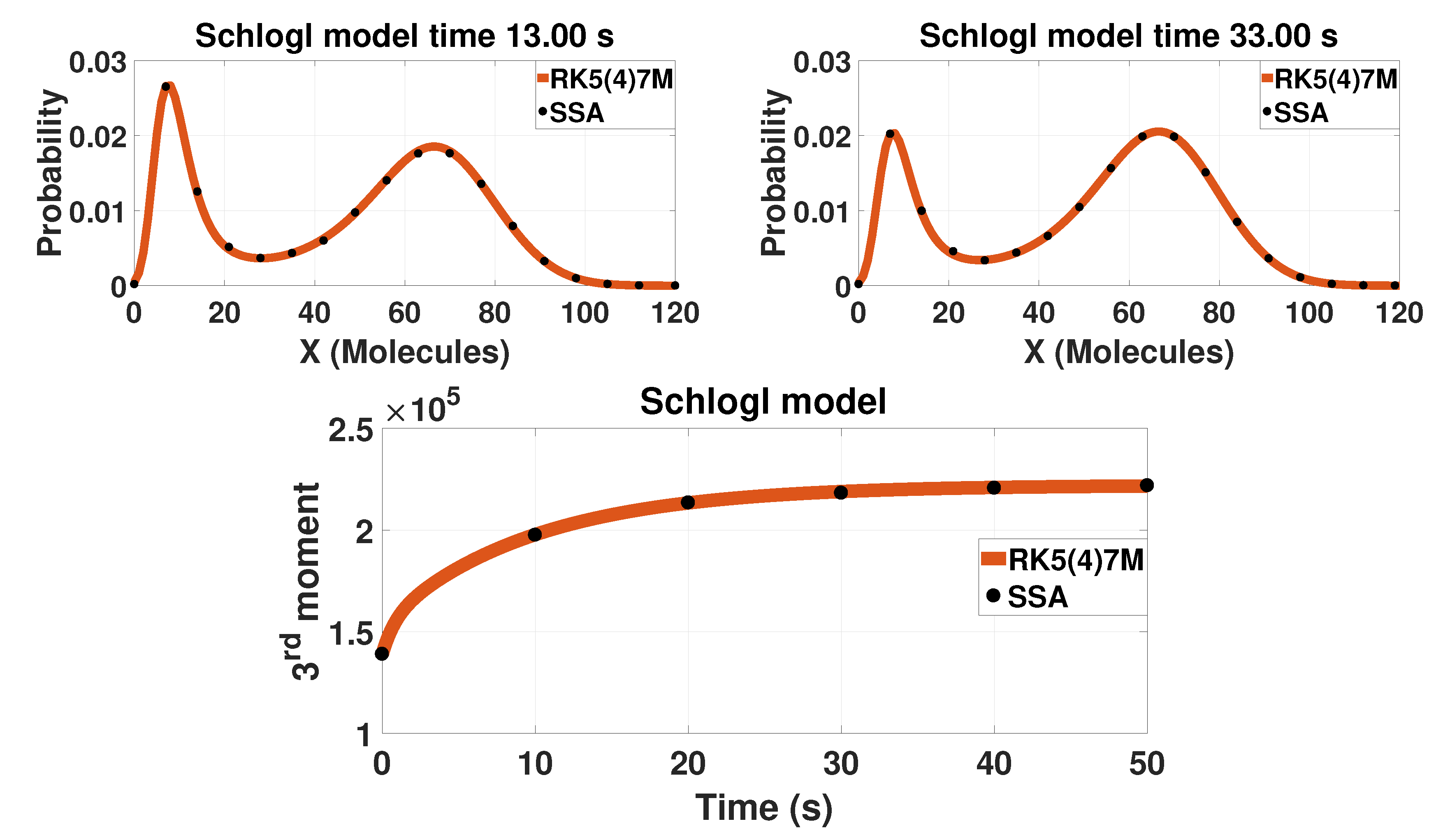
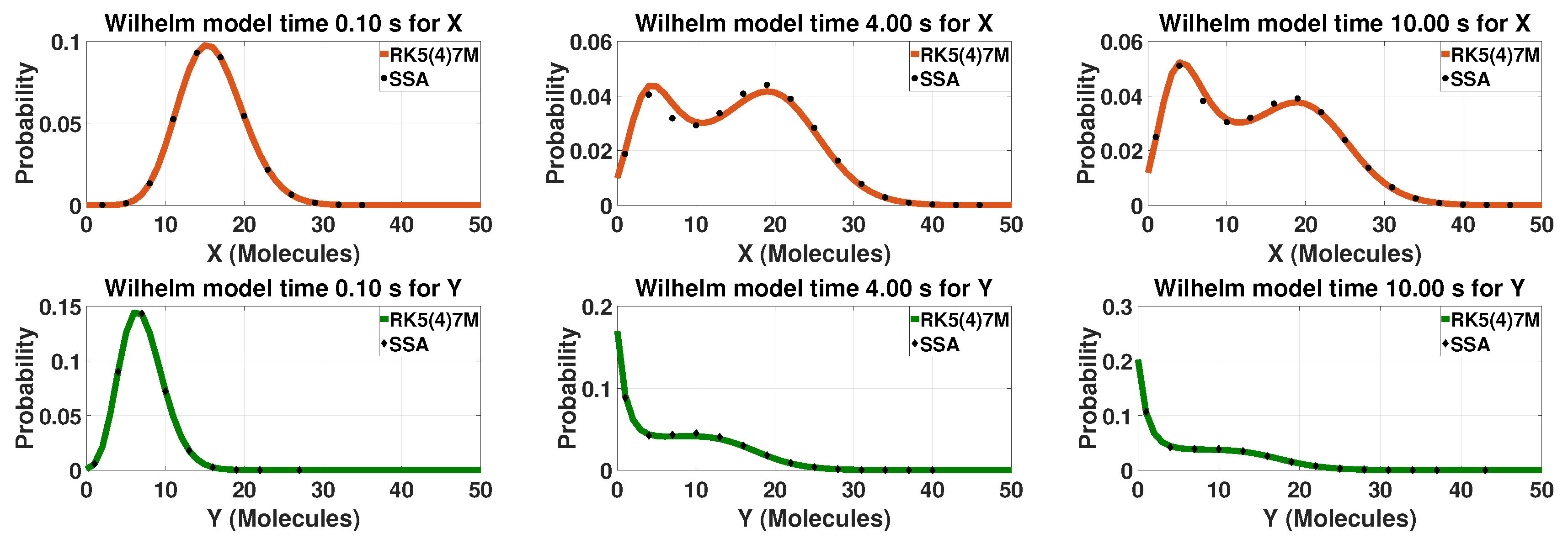
3.1. Multistable Systems: The Schlögl and Wilhelm Models
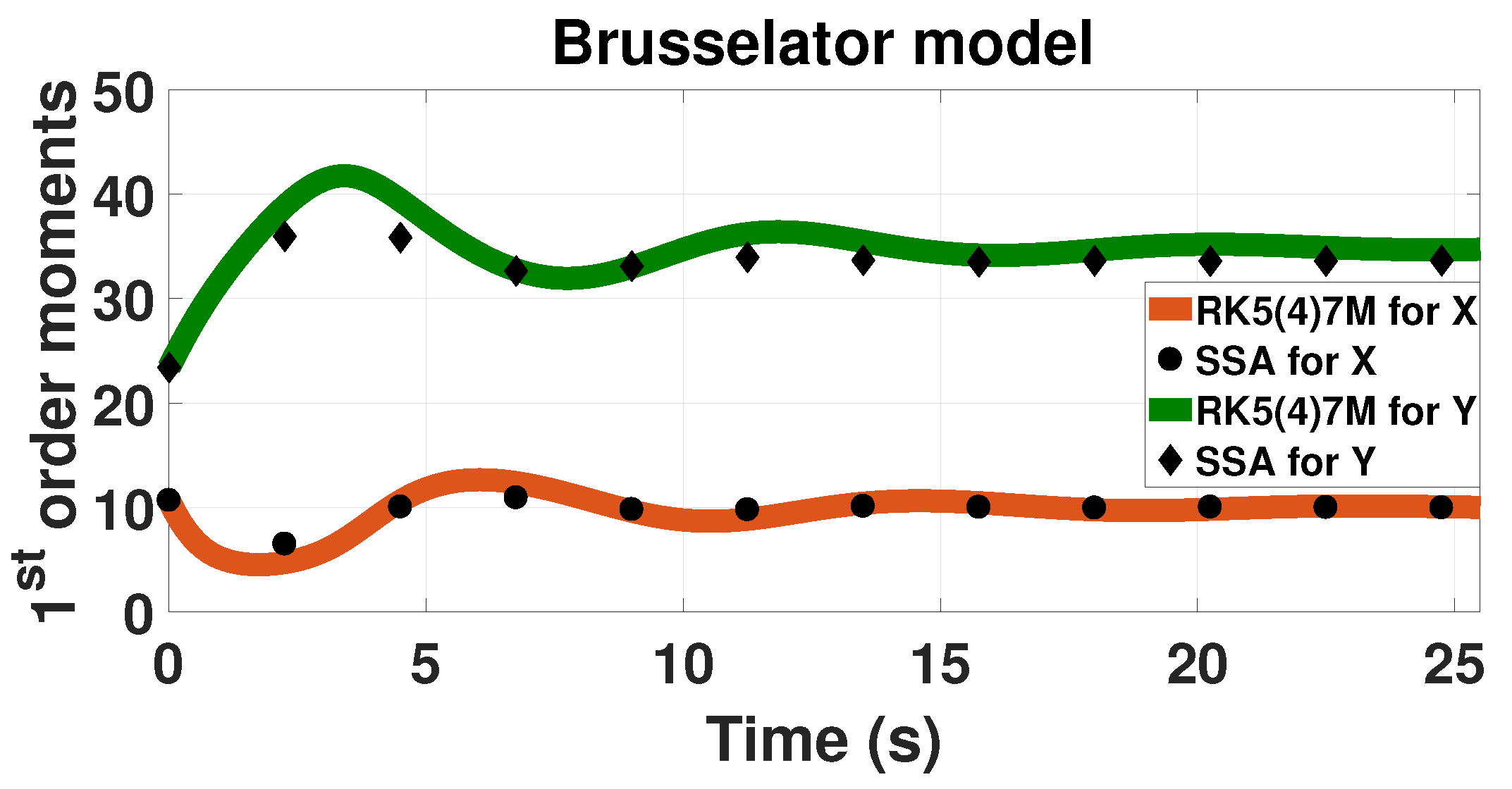
3.2. Oscillatory System: The Brusselator Model
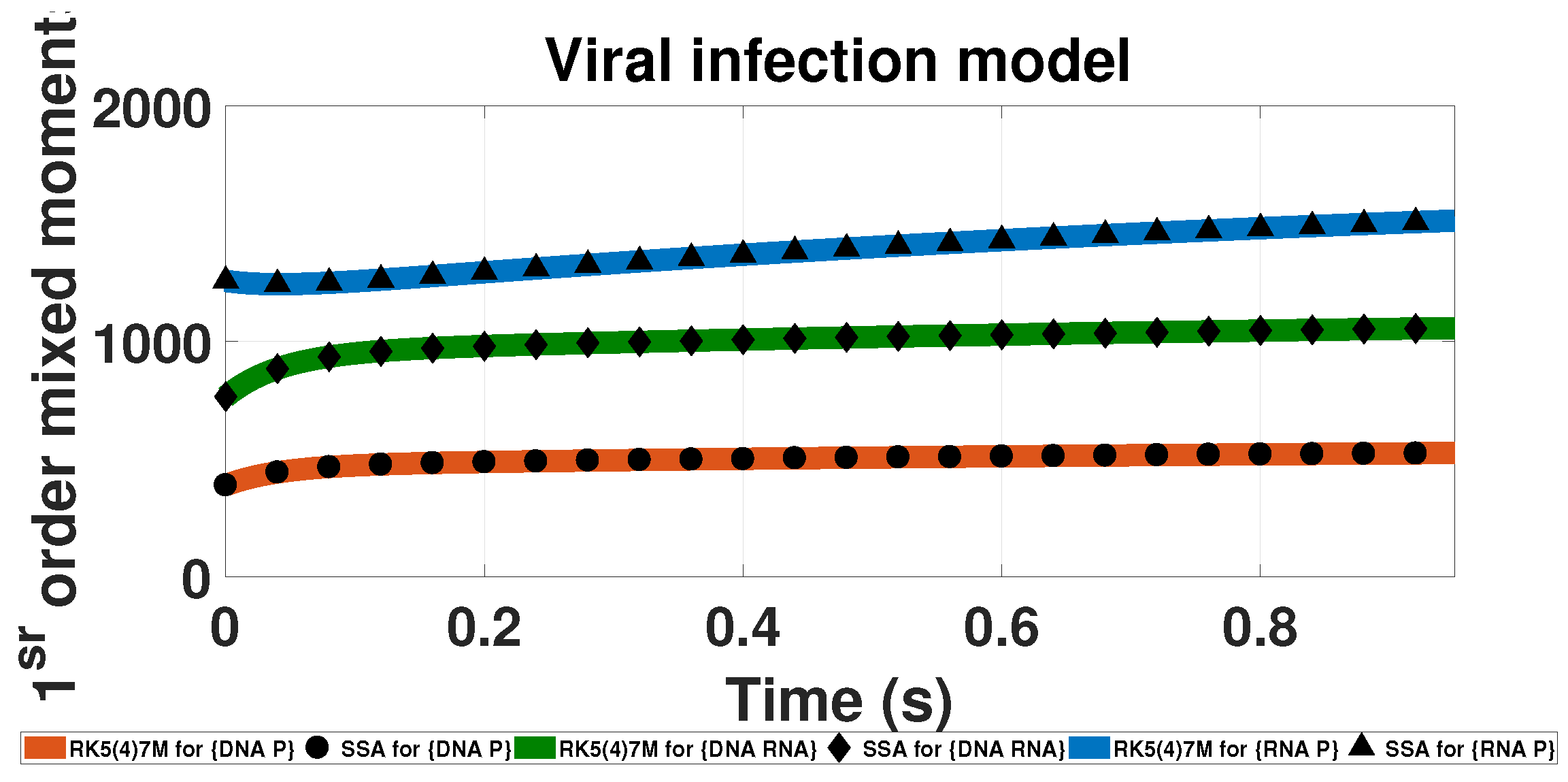
3.3. Multicomponent System: The Viral Infection Model
3.4. Computational Cost of LMEs
3.5. Dependence on State Space Size
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Appendix A. Inverse of Jacobian
Appendix B. Runge-Kutta
Appendix B.1. Runge-Kutta Tableau
| 1 | 2 | 3 | 4 | 5 | 6 | ||||
| i | |||||||||
| 1 | 0 | ||||||||
| 2 | 0 | 0 | |||||||
| 3 | |||||||||
| 4 | − | ||||||||
| 5 | − | − | − | − | |||||
| 6 | 1 | − | − | ||||||
| 7 | 1 | 0 | − | 0 | |||||
Appendix B.2. Runge-Kutta Algorithm
- Calculate vector and matrices A and as in [17]. These matrices depend only on the kinetic constants of the system and they are constant for all time points.
- Introduce the time step size h. The time point of this iteration is , where is the time point of the previous iteration.
- Increase the iteration counter by 1. For the rest of the algorithm the iteration step will be denoted by . At the first iteration, .
- Introduce the previous step (iteration n) Lagrange multipliers . At the first iteration, introduce the initial condition . The subscripts here refers to the time step and are applied on the whole Lagrange multiplier vector.
- Calculate the ’s coefficients from equation and the tableau (Appendix B.1). Each coefficient depends on the functional form F of the Lagrange multiplier equations () and on different set of Lagrange multipliers (). To account for the change of the Lagrange multipliers, there is a need for a subroutine to calculate .
- This is a proposed subroutine for
- (a)
- Calculate the augmented Lagrange multipliers for the specific , . The values of can be found in the tableau in Appendix B.1.
- (b)
- Calculate the lower () and higher-order moments () for the augmented Lagrange multipliers based on equation .
- (c)
- Calculate the inverse of the Jacobian, for the augmented multipliers as described in Appendix A.
- (d)
- Calculate the functional form of the Lagrange multiplier equations ().
- (e)
- Calculate based on equations .
- Repeat step 6 for all seven coefficients, . Its coefficient depends on all the previous coefficients, thus the calculation of each one needs to be in sequence and include the subroutine. For example, the calculations of step 6 should first performed for , then based on to be performed for , then based on to be performed for etc.
- Calculate the Lagrange multipliers for the current iteration , , based on equation and the tableau (Appendix B.1).
- Calculate the lower-order solution for the current iteration , , based on equation and the tableau (Appendix B.1).
- Check the accuracy of the solution by calculating the error :
- If the error is smaller than the wanted tolerance accept the solution and proceed to the next step. Otherwise, set a new time step size and go back to step 5 and re-perform the calculation with the new step size. s is used to adjust the step size based on the accuracy of the solution and is given by:
- Accept the solution of Lagrange multipliers for the current iteration () and current time point () as calculated at step 8.
- Set the new step size to and proceed to step 3 to calculate the solution for the rest of the time points until the final is reached.
- For any given time t, the probability distribution and its moments can be calculated based on the Lagrange multipliers () and equations & .
References
- Folger, H. Elements of Chemical Reaction Engineering, 4th ed.; Prentice Hall: Upper Saddle River, NJ, USA, 2016. [Google Scholar]
- Schnoerr, D.; Sanguinetti, G.; Grima, R. Approximation and inference methods for stochastic biochemical kinetics-A tutorial review. J. Phys. A 2017, 50, 093001. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Stochastic approach to chemical kinetics. J. Appl. Probab. 1967, 4, 413–478. [Google Scholar] [CrossRef]
- Oppenheim, I.; Shuler, K.E. Master Equations and Markov Processes. Phys. Rev. 1965, 138, B1007. [Google Scholar] [CrossRef]
- Kampen, N.V. Stochastic Processes in Physics and Chemistry, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2004; ISBN 978-0-444-89349-0. [Google Scholar]
- Gillespie, D.T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Gibson, M.A.; Bruck, J. Efficient Exact Stochastic Simulation of Chemical Systems with Many Species and Many Channels. J. Phys. Chem. A 2000, 104, 1876–1889. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Li, H.; Petzold, L. Efficient formulation of the stochastic simulation algorithm for chemically reacting systems. J. Chem. Phys. 2004, 121, 4059–4067. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T. Markov Processes, An Introduction for Physical Scientists; Academic Press: San Diego, CA, USA, 1992. [Google Scholar]
- Lakatos, E.; Ale, A.; Kirk, P.D.W.; Stumpf, M.P.H. Multivariate moment closure techniques for stochastic kinetic models. J. Chem. Phys. 2015, 143, 094107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munsky, B.; Khammash, M. The finite state projection algorithm for the solution of the chemical master equation. J. Chem. Phys. 2006, 124, 044104. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, T.; Huisinga, W. Solving the chemical master equation for monomolecular reaction systems analytically. J. Math. Bio. 2006, 54, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grima, R.; Schmidt, D.R.; Newman, T.J. Steady-state fluctuations of a genetic feedback loop: An exact solution. J. Chem. Phys. 2012, 137, 035104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, V.; Kaznessis, Y.N. An adaptive time step scheme for a system of stochastic differential equations with multiple multiplicative noise: Chemical Langevin equation, a proof of concept. J. Chem. Phys. 2008, 128, 014103. [Google Scholar] [CrossRef] [PubMed]
- Ale, A.; Kirk, P.; Stumpf, M.P.H. A general moment expansion method for stochastic kinetic models. J. Chem. Phys. 2013, 138, 174101. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulos, V.; Kaznessis, Y.N. Analytical Derivation of Moment Equations in Stochastic Chemical Kinetics. Chem. Eng. Sci. 2011, 66, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Smadbeck, P.; Kaznessis, Y.N. Efficient Moment Matrix Generation for Arbitrary Chemical Networks. Chem. Eng. Sci. 2012, 84, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, C. Moment-closure approximations for mass-action models. IET Syst. Biol. 2009, 3, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Grima, R. A study of the accuracy of moment-closure approximations for stochastic chemical kinetics. J. Chem. phys. 2012, 136, 154105. [Google Scholar] [CrossRef] [PubMed]
- Constantino, P.H.; Vlysidis, M.; Smadbeck, P.; Kaznessis, Y.N. Modeling stochasticity in biochemical reaction networks. J. Phys. D Appl. Phys. 2016, 49, 093001. [Google Scholar] [CrossRef]
- Milner, P.; Gillespie, C.S.; Wilkinson, D.J. Moment closure based parameter inference of stochastic kinetic models. Stat. Comput. 2013, 23, 287–295. [Google Scholar] [CrossRef]
- Smadbeck, P.; Kaznessis, Y.N. A closure scheme for chemical master equations. Proc. Natl. Acad. Sci. USA 2013, 110, 14261–14265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechner, C.; Ruess, J.; Krenn, P.; Pelet, S.; Peter, M.; Lygeros, J.; Koeppl, H. Moment-based inference predicts bimodality in transient gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 8340–8345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Hespanha, J. Moment closure techniques for stochastic models in population biology. In Proceedings of the 2006 American Control Conference, Minneapolis, MN, USA, 14–16 June 2006. [Google Scholar]
- Krishnarajah, I.; Cook, A.; Marion, G.; Gibson, G. Novel moment closure approximations in stochastic epidemics. Bull. Math. Biol. 2005, 67, 855–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnoerr, D.; Sanguinetti, G.; Grima, R. Comparison of different moment-closure approximations for stochastic chemical kinetics. J. Chem. Phys. 2015. [Google Scholar] [CrossRef] [PubMed]
- Goutsias, J.; Jenkinson, G. Markovian dynamics on complex reaction networks. Phys. Rep. 2013, 2, 199–264. [Google Scholar] [CrossRef]
- Kapur, J. Maximum-Entropy Models in Science and Engineering; Wiley Eastern Ltd.: Darya Ganj, New Delhi, 1989. [Google Scholar]
- Vlysidis, M.; Constantino, P.H.; Kaznessis, Y.N. ZI-Closure Scheme: A Method to Solve and Study Stochastic Reaction Networks. In Stochastic Processes, Multiscale Modeling, and Numerical Methods for Computational Cellular Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 159–174. ISBN 978-3-319-62627-7. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Sutter, T.; Sutter, D.; Esfahani, P.M.; Lygeros, J. Generalized Maximum Entropy Estimation. arXiv 2004, arXiv:1708.07311. Available online: https://arxiv.org/abs/1708.07311 (accessed on 6 September 2018).
- Dormand, J.; Prince, P. A family of embedded Runge-Kutta formulae. J. Comput. Appl. Math. 1980, 6, 19–26. [Google Scholar] [CrossRef]
- Schlogl, F. On Thermodynamics Near a Steady State. Z. Physik 1971, 458, 446–458. [Google Scholar] [CrossRef]
- Wilhelm, T. The smallest chemical reaction system with bistability. BMC Syst. Biol. 2009, 3, 90. [Google Scholar] [CrossRef] [PubMed]
- Lefever, R.; Nicolis, G. Chemical instabilities and sustained oscillations. J. Theor. Biol. 1971, 30, 267–284. [Google Scholar] [CrossRef]
- Lavenda, B.; Nicolis, G.; Herschkowitz-Kaufman, M. Chemical instabilities and relaxation oscillations. J. Theor. Biol. 1971, 32, 283–292. [Google Scholar] [CrossRef]
- Haseltine, E.L.; Rawlings, J.B. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J. Chem. Phys. 2002, 117, 6959–6969. [Google Scholar] [CrossRef] [Green Version]
- Goutsias, J. Quasiequilibrium approximation of fast reaction kinetics in stochastic biochemical systems. J. Chem. Phys. 2005, 122, 184102. [Google Scholar] [CrossRef] [PubMed]
- Vlysidis, M.; Kaznessis, Y.N. A linearization method for probability moment equations. Comput. Chem. Eng. 2018, 112, 1–5. [Google Scholar] [CrossRef]
- Munkhammar, J.; Mattsson, L.; Rydén, J. Polynomial probability distribution estimation using the method of moments. PLoS ONE 2017, 12, e0174573. [Google Scholar] [CrossRef] [PubMed]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Gardner, T.S.; Cantor, C.R.; Collins, J.J. Construction of a genetic toggle switch in Escherichia coli. Nature 2000, 403, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Angeli, D.; Ferrell, J.E.; Sontag, E.D.; Sontag, E.D. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl. Acad. Sci. USA 2004, 101, 1822–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artyomov, M.N.; Das, J.; Kardar, M.; Chakraborty, A.K. Purely stochastic binary decisions in cell signaling models without underlying deterministic bistabilities. Proc. Natl. Acad. Sci. USA 2007, 104, 18958–18963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlysidis, M.; Schiek, A.C.; Kaznessis, Y.N. ZICS: An Application for Calculating the Stationary Probability Distribution of Stochastic Reaction Networks. arXiv 2018, arXiv:1806.06428. Available online: https://arxiv.org/abs/1806.06428 (accessed on 6 September 2018).
- Constantino, P.H.; Kaznessis, Y.N. Maximum entropy prediction of non-equilibrium stationary distributions for stochastic reaction networks with oscillatory dynamics. Chem. Eng. Sci. 2017, 171, 139–148. [Google Scholar] [CrossRef]
- Peleš, S.; Munsky, B.; Khammash, M. Reduction and solution of the chemical master equation using time scale separation and finite state projection. J. Chem. Phys. 2006, 125, 204104. [Google Scholar] [CrossRef] [PubMed]
- Golub, G.H.; Van Loan, C.F. Matrix Computations, 4th ed.; The Johns Hopkins University Press: Baltimore, MD, USA, 2013; ISBN 978-1-4214-0794-4. [Google Scholar]
- Butcher, J.C. A stability property of implicit Runge-Kutta methods. BIT 1975, 15, 358–361. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial Moments | ||||
|---|---|---|---|---|
| Models | Reactions | Kinetic Constants | First-Order | Second-Order |
| Schlögl | ||||
| Wilhelm | ||||
| Brusselator | ||||
| Viral Infection* | ||||
| Network | Average Error (%) of | Time per Step (CPU s) | ||||
|---|---|---|---|---|---|---|
| 1st-Orde Moments | 2nd-Order Moments | LMEs | SSA | Time Ratio | ||
| Schlögl | 0.05 | 0.22 | 0.22 | 0.02 | 419.2 | 20,960 |
| Wilhelm | 0.61 | 0.56 | 0.71 | 0.15 | 732.75 | 4,885 |
| Brusselator | 9.90 | 2.81 | 6.07 | 0.74 | 68.05 | 92 |
| Viral Infection | 0.43 | 0.01 | 0.02 | 8.97 | 64.96 | 7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlysidis, M.; Kaznessis, Y.N. Solving Stochastic Reaction Networks with Maximum Entropy Lagrange Multipliers. Entropy 2018, 20, 700. https://doi.org/10.3390/e20090700
Vlysidis M, Kaznessis YN. Solving Stochastic Reaction Networks with Maximum Entropy Lagrange Multipliers. Entropy. 2018; 20(9):700. https://doi.org/10.3390/e20090700
Chicago/Turabian StyleVlysidis, Michail, and Yiannis N. Kaznessis. 2018. "Solving Stochastic Reaction Networks with Maximum Entropy Lagrange Multipliers" Entropy 20, no. 9: 700. https://doi.org/10.3390/e20090700
APA StyleVlysidis, M., & Kaznessis, Y. N. (2018). Solving Stochastic Reaction Networks with Maximum Entropy Lagrange Multipliers. Entropy, 20(9), 700. https://doi.org/10.3390/e20090700