Critical Size of Secondary Nuclei Determined via Nucleation Theorem Reveals Selective Nucleation in Three-Component Co-Crystals



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
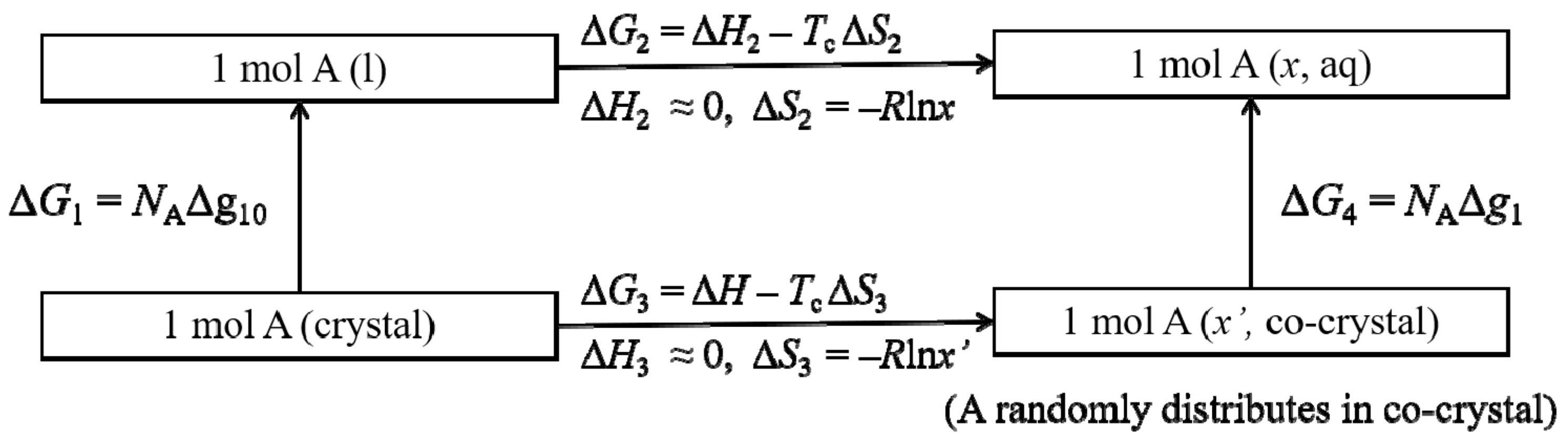{kind=link}
Abstract
:1. Introduction
2. Theory
3. Experimental Section
3.1. Sample Preparation
3.2. Characterization
4. Results and Discussion
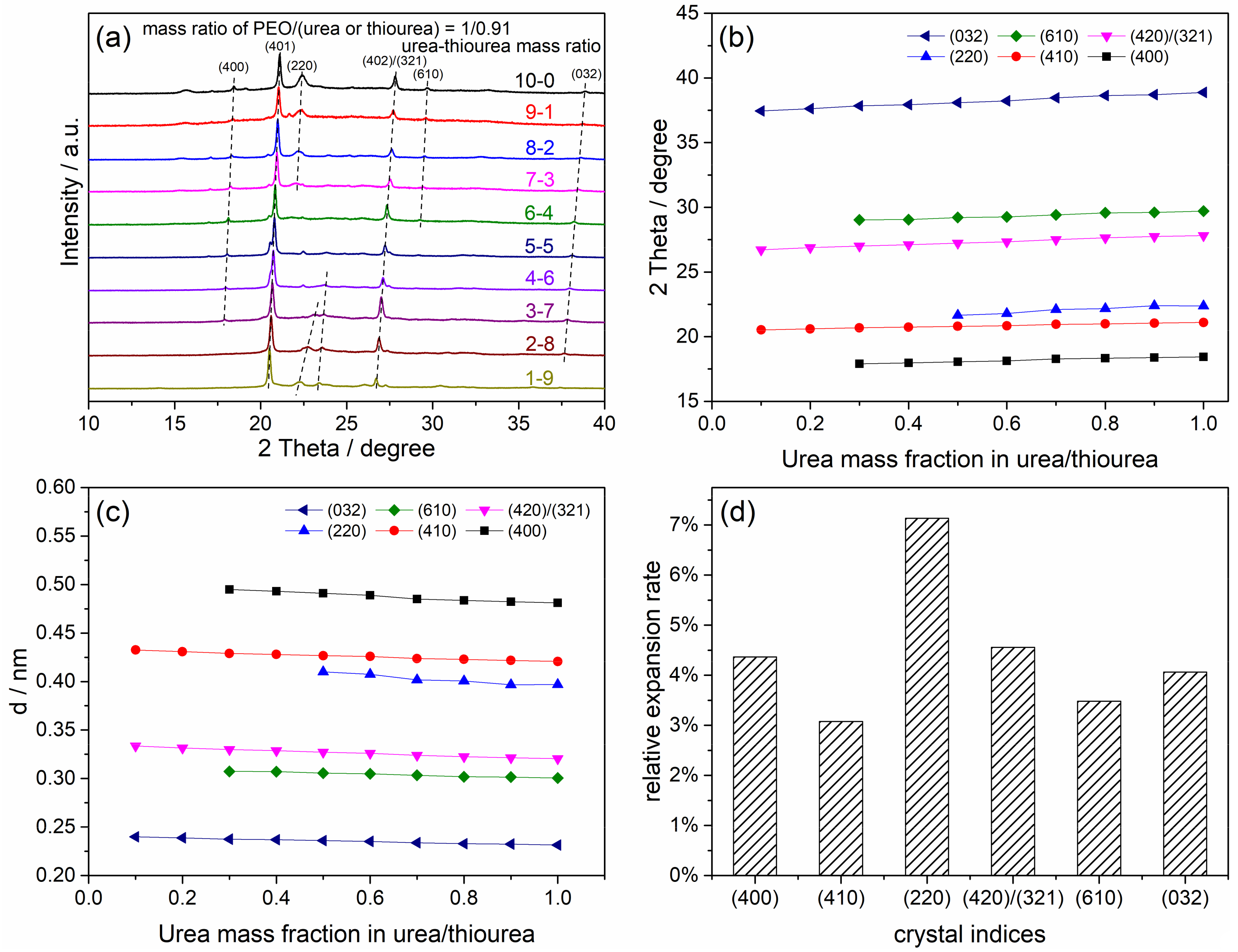
4.1. Crystalline Structure of the Peo/Urea/Thiourea Ternary Inclusion Compounds
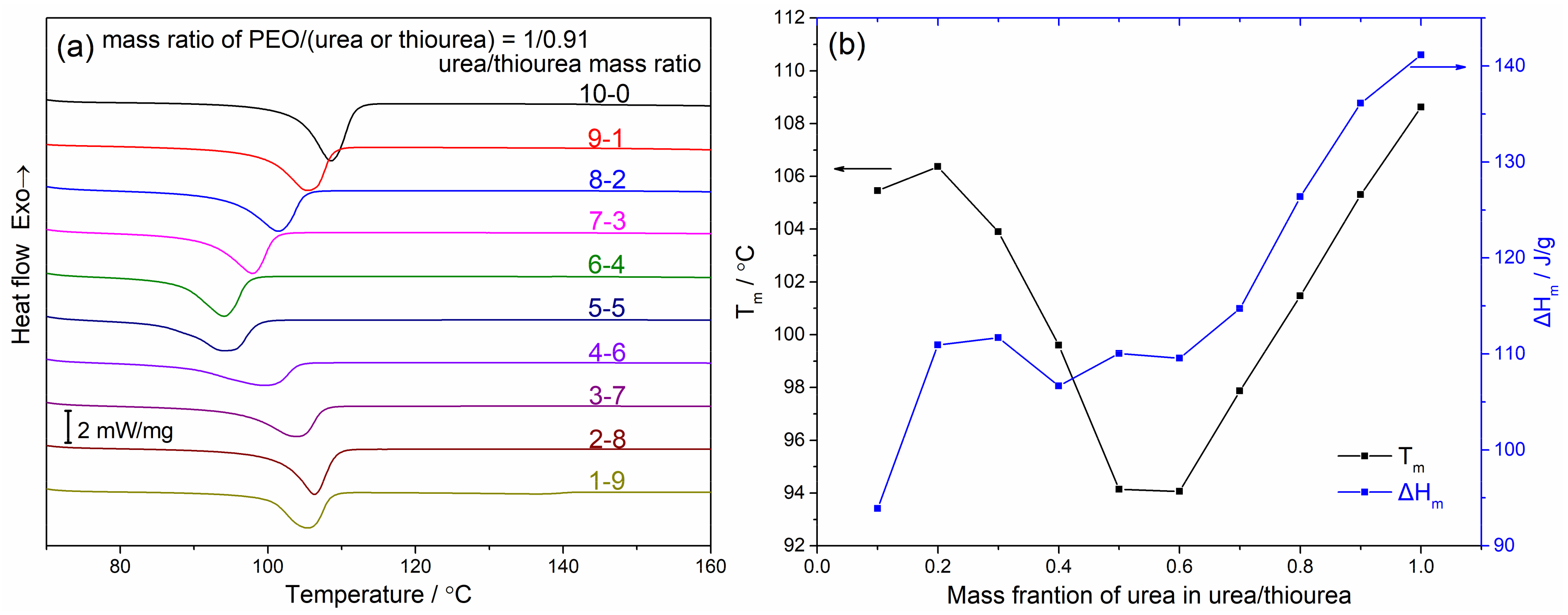
4.2. Thermodynamic Stability of the PEO/Urea/Thiourea Ternary Inclusion Compounds
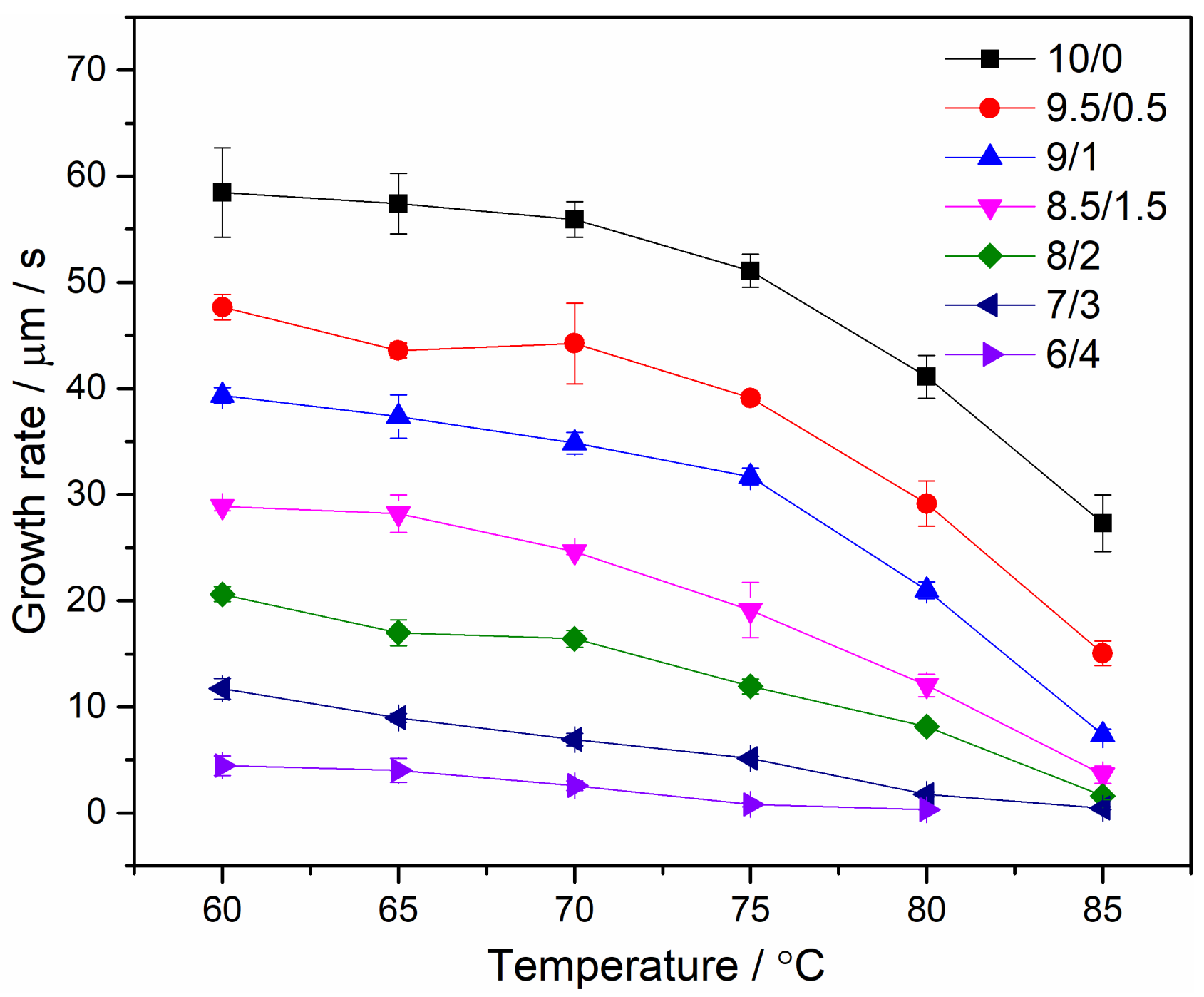
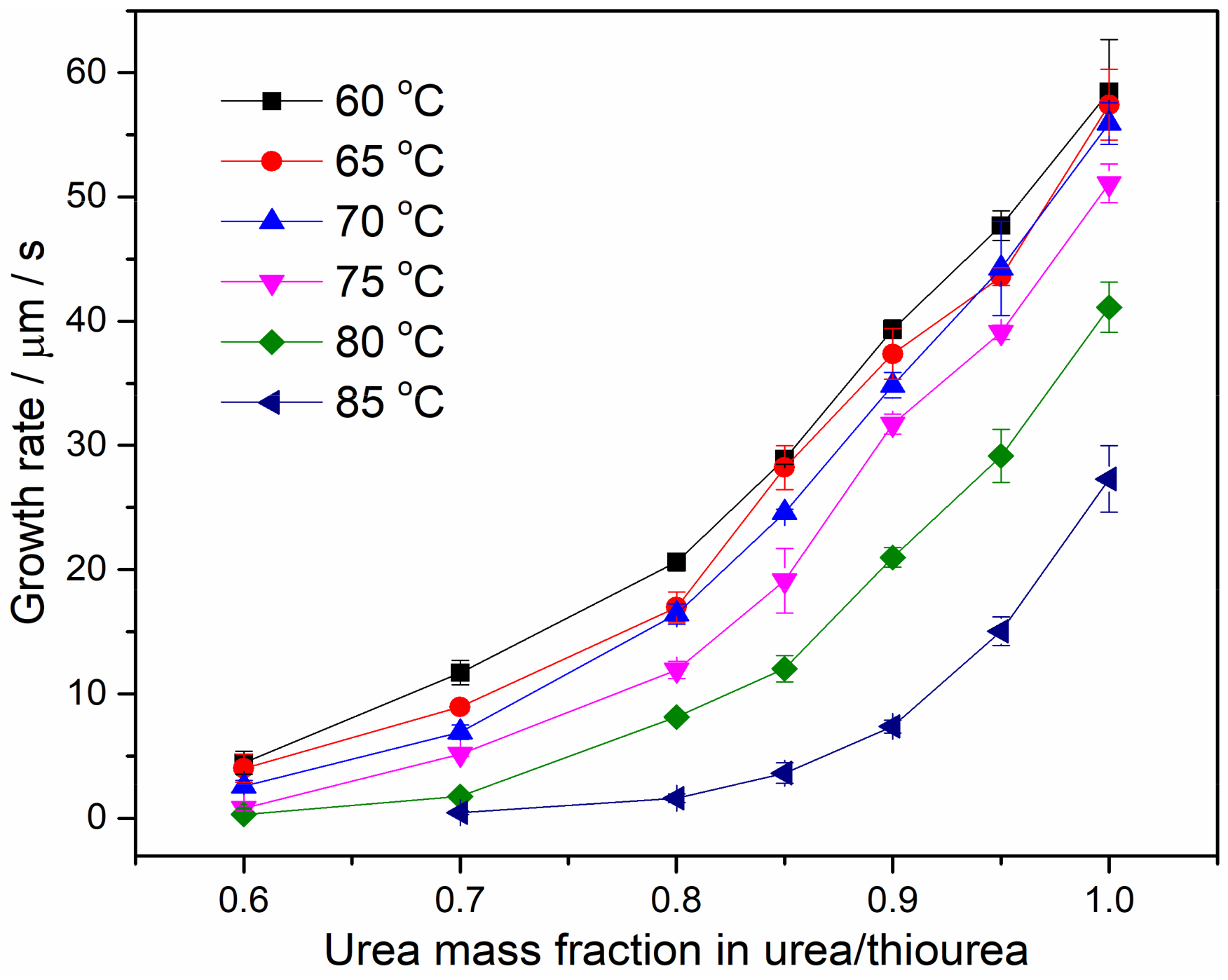
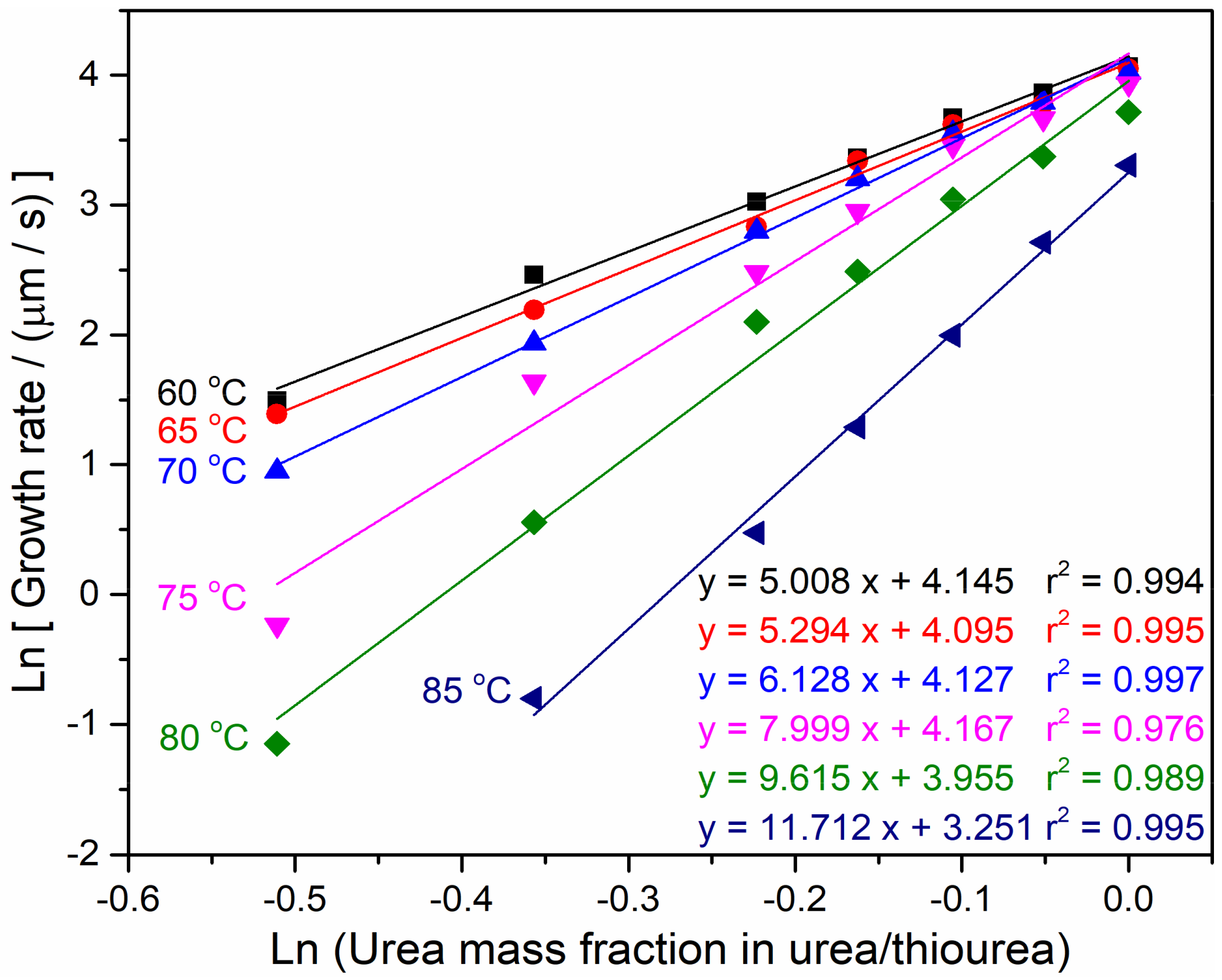
4.3. Spherulite Morphologies and Radial Growth Rate of PEO/Urea/Thiourea IC
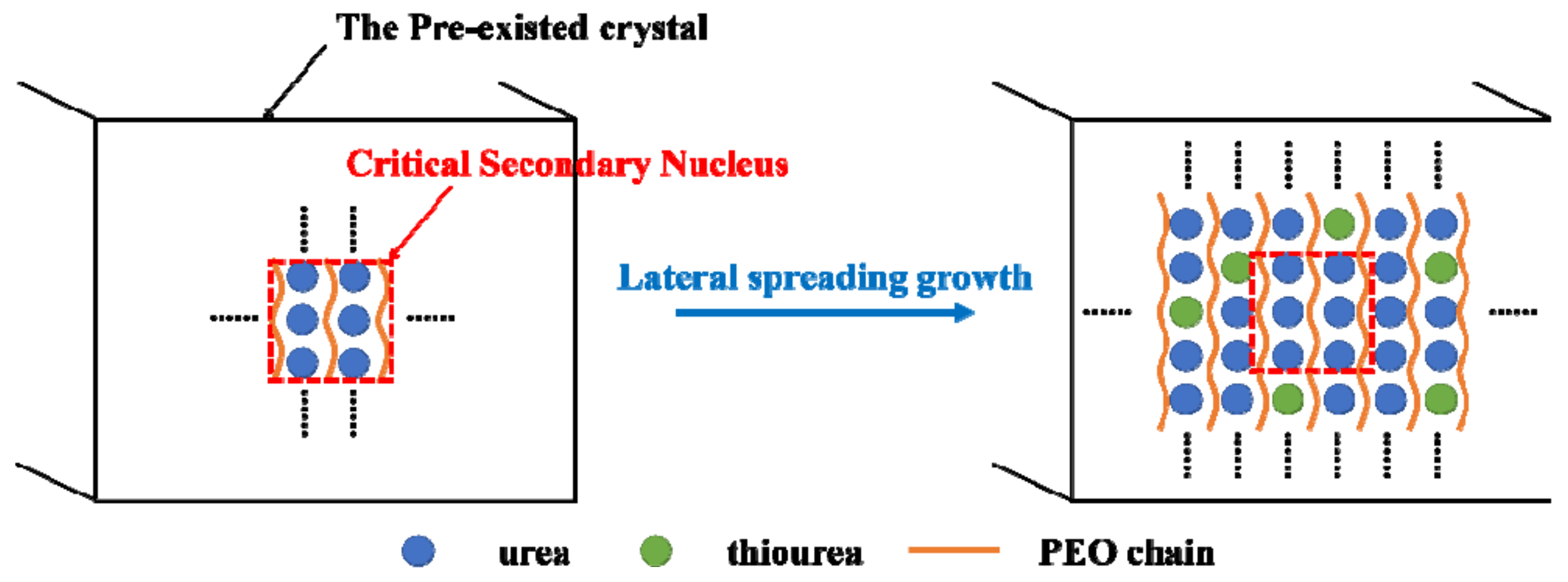
4.4. Critical Size of the Secondary Nucleus of PEO/Urea/Thiourea IC
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schick, C.; Androsch, R.; Schmelzer, J.W.P. Homogeneous crystal nucleation in polymers. J. Phys. Condens. Matter 2017, 29, 453002. [Google Scholar] [CrossRef]
- Schmelzer, J.; Röpke, G.; Priezzhev, V.B. Nucleation Theory and Applications; Wiley Online Library: Hoboken, NJ, USA, 2005; Volume 76. [Google Scholar]
- Xu, J.; Zhang, S.; Guo, B. Insights from polymer crystallization: Chirality, recognition and competition. Chin. Chem. Lett. 2017, 28, 2092–2098. [Google Scholar] [CrossRef]
- Mutaftschiev, B. The Atomistic Nature of Crystal Growth; Springer Science & Business Media: Berlin, Germany, 2001; Volume 43. [Google Scholar]
- Volmer, M.; Weber, A. Keimbildung in übersättigten Gebilden. Z. Phys. Chem. 1926, 119, 277–301. [Google Scholar] [CrossRef]
- Becker, R.; Döring, W. Kinetische Behandlung der Keimbildung in übersättigten Dämpfen. Ann. Phys. 1935, 416, 719–752. [Google Scholar] [CrossRef]
- Turnbull, D.; Fisher, J.C. Rate of Nucleation in Condensed Systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Armitstead, K.; Goldbeck-Wood, G.; Keller, A. Polymer crystallization theories. In Macromolecules: Synthesis, Order and Advanced Properties; Springer: Berlin/Heidelberg, Germany, 1992; pp. 219–312. [Google Scholar]
- Zhang, M.C.; Guo, B.; Xu, J. A Review on Polymer Crystallization Theories. Crystals 2017, 7, 4. [Google Scholar] [CrossRef]
- Xu, K.-L.; Guo, B.-H.; Reiter, R.; Reiter, G.; Xu, J. Simulation of secondary nucleation of polymer crystallization via a model of microscopic kinetics. Chin. Chem. Lett. 2015, 26, 1105–1108. [Google Scholar] [CrossRef]
- Šarić, A.; Michaels, T.C.T.; Zaccone, A.; Knowles, T.P.J.; Frenkel, D. Kinetics of spontaneous filament nucleation via oligomers: Insights from theory and simulation. J. Chem. Phys. 2016, 145, 211926. [Google Scholar] [CrossRef]
- Kashchiev, D. On the relation between nucleation work, nucleus size, and nucleation rate. J. Chem. Phys. 1982, 76, 5098–5102. [Google Scholar] [CrossRef]
- Oxtoby, D.W.; Kashchiev, D. A general relation between the nucleation work and the size of the nucleus in multicomponent nucleation. J. Chem. Phys. 1994, 100, 7665–7671. [Google Scholar] [CrossRef]
- Kashchiev, D. Forms and applications of the nucleation theorem. J. Chem. Phys. 2006, 125, 014502. [Google Scholar] [CrossRef] [PubMed]
- Ford, I.J. Nucleation theorems, the statistical mechanics of molecular clusters, and a revision of classical nucleation theory. Phys. Rev. E 1997, 56, 5615–5629. [Google Scholar] [CrossRef] [Green Version]
- Ford, I.J. Statistical mechanics of nucleation: A review. Proc. Inst. Mech. Eng. Part C: J. Mech. Eng. Sci. 2004, 218, 883–899. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, S.; Shi, J.; Guo, B.; Xu, J. Study of the Crystal Growth Mechanism and Critical Secondary Nucleus Size of Poly (Ethylene oxide)/Urea Inclusion Compound. Cryst. Growth Des. 2019, 19, 3834–3842. [Google Scholar] [CrossRef]
- Tadokoro, H.; Yoshihara, T.; Chatani, Y.; Murahashi, S. A preliminary report of structural studies on polyethylene oxide-urea complex. J. Polym. Sci. Part B Polym. Lett. 1964, 2, 363–368. [Google Scholar] [CrossRef]
- Suehiro, K.; Nagano, Y. Structural studies on molecular complexes of polyethers, 1. Urea-ethylene oxide oligomer complexes. Makromol. Chem. 1983, 184, 669–674. [Google Scholar] [CrossRef]
- Chenite, A.; Brisse, F. Structure and conformation of poly (ethylene oxide), PEO, in the trigonal form of the PEO-urea complex at 173 K. Macromolecules 1991, 24, 2221–2225. [Google Scholar] [CrossRef]
- Ye, H.-M.; Peng, M.; Xu, J.; Guo, B.-H.; Chen, Q.; Yun, T.-L.; Ma, H. Conformation transition and molecular mobility of isolated poly (Ethylene oxide) chains confined in urea nanochannels. Polymer 2007, 48, 7364–7373. [Google Scholar] [CrossRef]
- Liu, Y.; Pellerin, C. Highly oriented electrospun fibers of self-assembled inclusion complexes of poly (Ethylene oxide) and urea. Macromolecules 2006, 39, 8886–8888. [Google Scholar] [CrossRef]
- Liu, Y.; Antaya, H.; Pellerin, C. Characterization of the stable and metastable poly (Ethylene oxide)-urea complexes in electrospun fibers. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 1903–1913. [Google Scholar] [CrossRef]
- Gao, Y.; Yao, S.; Ye, H.; Guo, B.; Xu, J. Orientation of polymer chains in spherulites of poly (Ethylene oxide)-urea inclusion compounds. Polymer 2017, 130, 209–217. [Google Scholar] [CrossRef]
- Lin, D.-W.; Zhong, Z.; Tang, Y.-R.; Gao, Y.; He, Y.-N.; Guo, B.-H.; Xu, J. Morphology and crystalline structure of inclusion compounds formed between poly (Ethylene glycol) and urea. Chin. J. Polym. Sci. 2014, 32, 1234–1242. [Google Scholar] [CrossRef]
- Zhong, Z.; Yang, X.; Wang, B.-H.; Yao, Y.-F.; Guo, B.; Yu, L.; Huang, Y.; Xu, J. Solvent-polymer guest exchange in a carbamazepine inclusion complex: Structure, kinetics and implication for guest selection. CrystEngComm 2019, 21, 2164–2173. [Google Scholar] [CrossRef]
- Liu, Y.; Pellerin, C. Stability and phase behavior of the poly (Ethylene oxide)–urea complexes prepared by electrospinning. Polymer 2009, 50, 2601–2607. [Google Scholar] [CrossRef]
- Ye, H.-M.; Hong, L.-T.; Gao, Y.; Xu, J. Isomorphism in ternary complex: Poly (Ethylene oxide), urea and thiourea. Chin. Chem. Lett. 2017, 28, 888–892. [Google Scholar] [CrossRef]
- Oxtoby, D.W. Nucleation of First-Order Phase Transitions. Acc. Chem. Res. 1998, 31, 91–97. [Google Scholar] [CrossRef]
- Sear, R.P. Nucleation: Theory and applications to protein solutions and colloidal suspensions. J. Phys. Condens. Matter 2007, 19, 033101. [Google Scholar] [CrossRef]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Nucleation of Crystals from Solution: Classical and Two-Step Models. Acc. Chem. Res. 2009, 42, 621–629. [Google Scholar] [CrossRef]
- Vekilov, P.G. Nucleation. Cryst. Growth Des. 2010, 10, 5007–5019. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P. Comments on the Nucleation Theorem. J. Colloid Interface Sci. 2001, 242, 354–372. [Google Scholar] [CrossRef]
- Ye, H.-M.; Wang, R.-D.; Liu, J.; Xu, J.; Guo, B.-H. Isomorphism in Poly (butylene succinate-co-butylene fumarate) and Its Application as Polymeric Nucleating Agent for Poly (Butylene succinate). Macromolecules 2012, 45, 5667–5675. [Google Scholar] [CrossRef]
- Pérez-Camargo, R.A.; Arandia, I.; Safari, M.; Cavallo, D.; Lotti, N.; Soccio, M.; Müller, A.J. Crystallization of isodimorphic aliphatic random copolyesters: Pseudo-eutectic behavior and double-crystalline materials. Eur. Polym. J. 2018, 101, 233–247. [Google Scholar] [CrossRef]
- Ye, H.-M.; Wang, J.; Wang, C.-S.; Li, H.-F. Unique Isodimorphism of Poly (decamethylene succinate-ran-decamethylene fumarate): Large Pseudoeutectic Region and Fantastic Crystallization/Melting Behavior. Macromolecules 2019, 52, 1447–1457. [Google Scholar] [CrossRef]
- Liu, Y.; Antaya, H.; Pellerin, C. Structure and Phase Behavior of the Poly (Ethylene oxide)—Thiourea Complex Prepared by Electrospinning. J. Phys. Chem. B 2010, 114, 2373–2378. [Google Scholar] [CrossRef]
- Ye, H.-M.; Liu, K.-S.; Zhou, Q. Inclusion complex formed between poly (Ethylene oxide) and thiourea. Colloids Surf. Physicochem. Eng. Asp. 2015, 467, 251–258. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Guo, B.; Xu, J. Critical Size of Secondary Nuclei Determined via Nucleation Theorem Reveals Selective Nucleation in Three-Component Co-Crystals. Entropy 2019, 21, 1032. https://doi.org/10.3390/e21111032
Gao Y, Guo B, Xu J. Critical Size of Secondary Nuclei Determined via Nucleation Theorem Reveals Selective Nucleation in Three-Component Co-Crystals. Entropy. 2019; 21(11):1032. https://doi.org/10.3390/e21111032
Chicago/Turabian StyleGao, Yang, Baohua Guo, and Jun Xu. 2019. "Critical Size of Secondary Nuclei Determined via Nucleation Theorem Reveals Selective Nucleation in Three-Component Co-Crystals" Entropy 21, no. 11: 1032. https://doi.org/10.3390/e21111032
APA StyleGao, Y., Guo, B., & Xu, J. (2019). Critical Size of Secondary Nuclei Determined via Nucleation Theorem Reveals Selective Nucleation in Three-Component Co-Crystals. Entropy, 21(11), 1032. https://doi.org/10.3390/e21111032