A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies


Abstract
:1. Introduction
2. Results
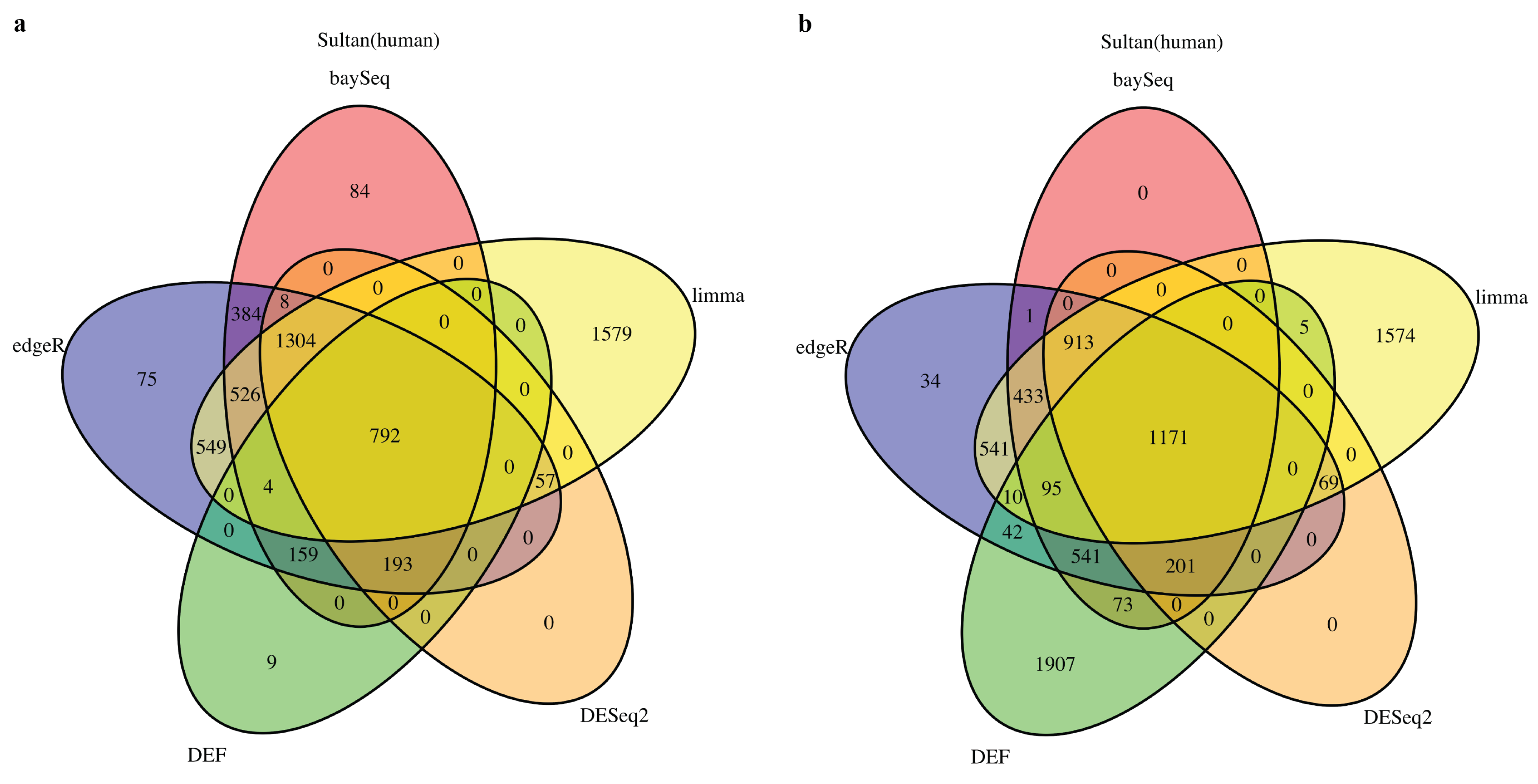
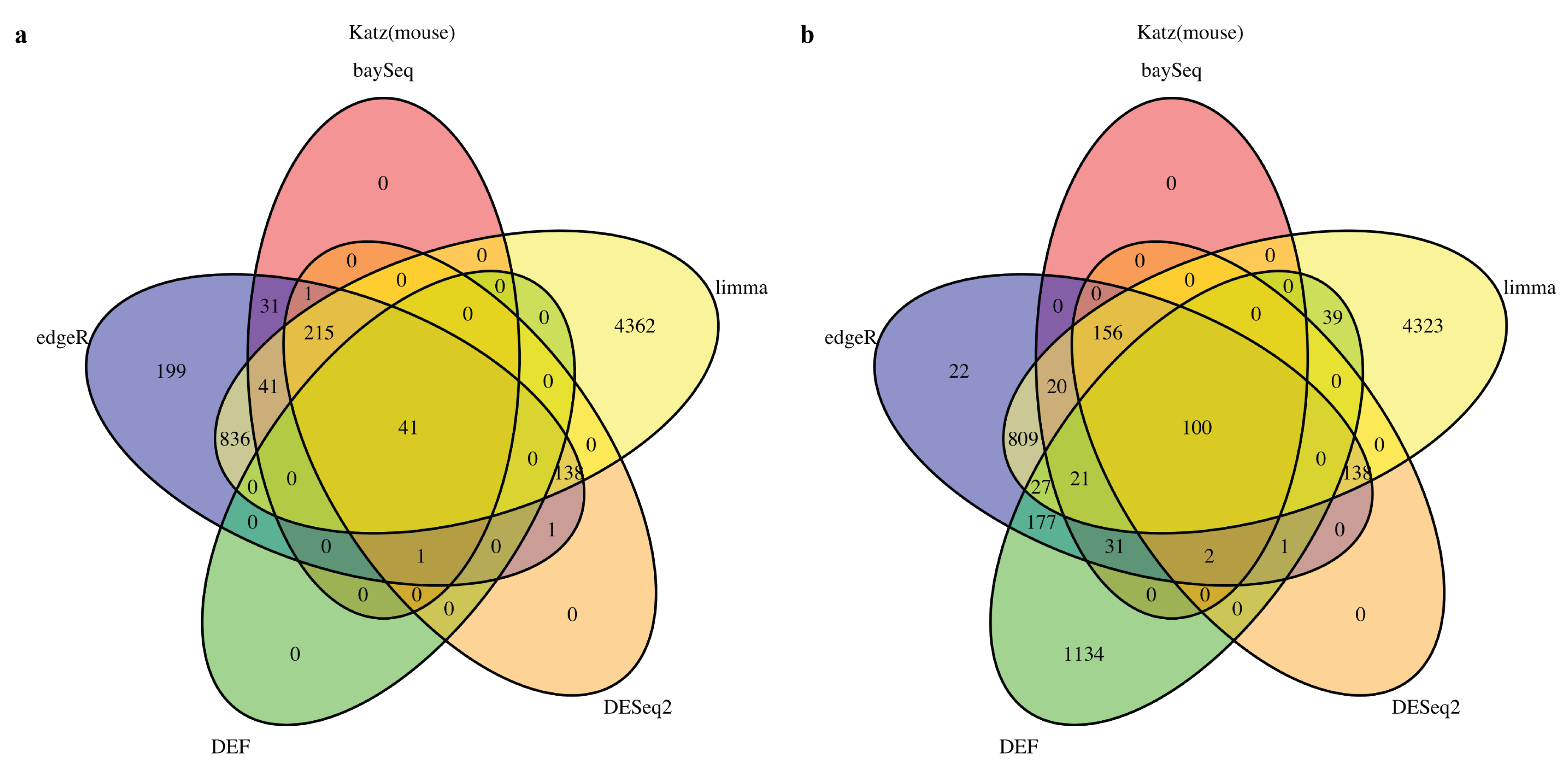
2.1. DEF Shares Many Genes with Limma, DESeq2, baySeq and edgeR
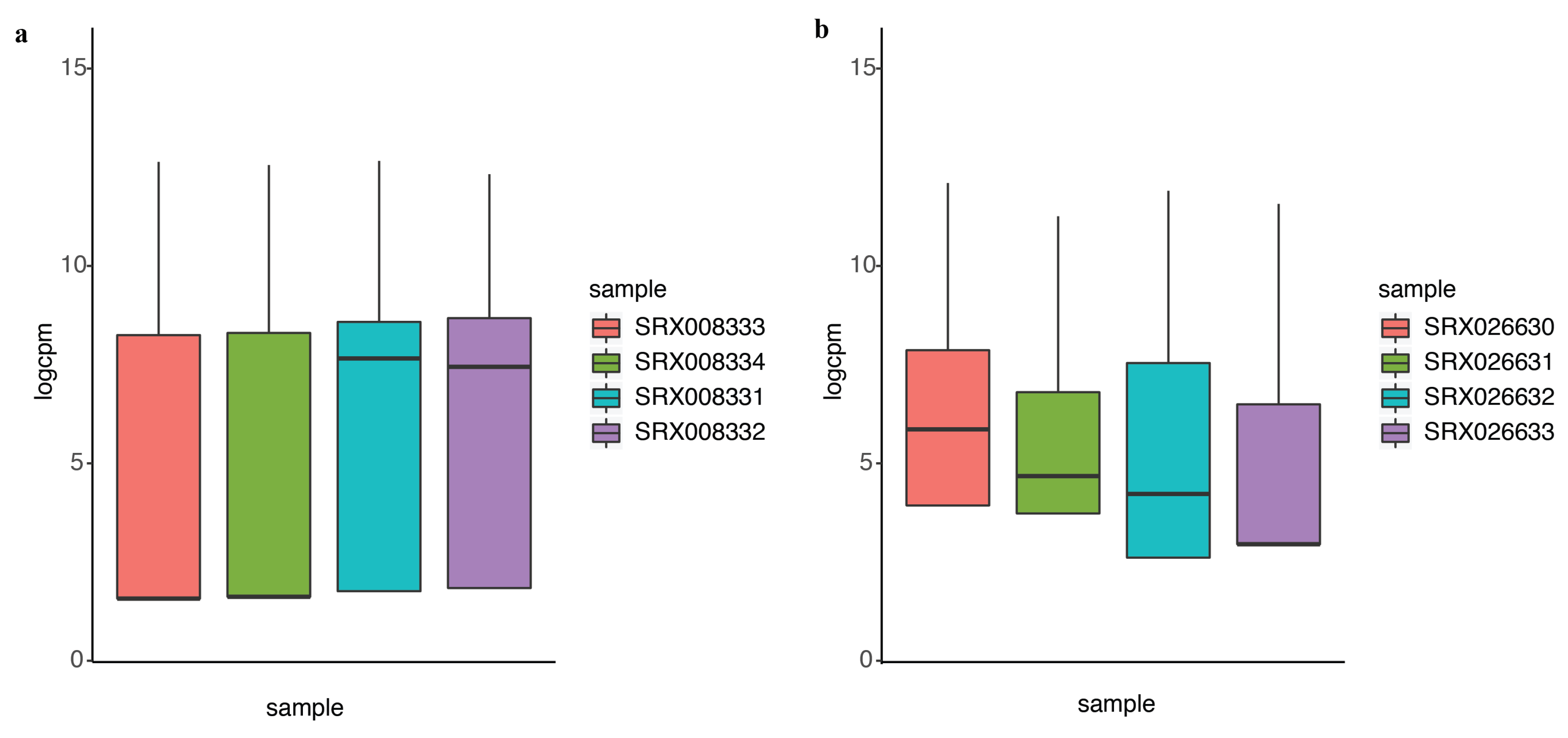
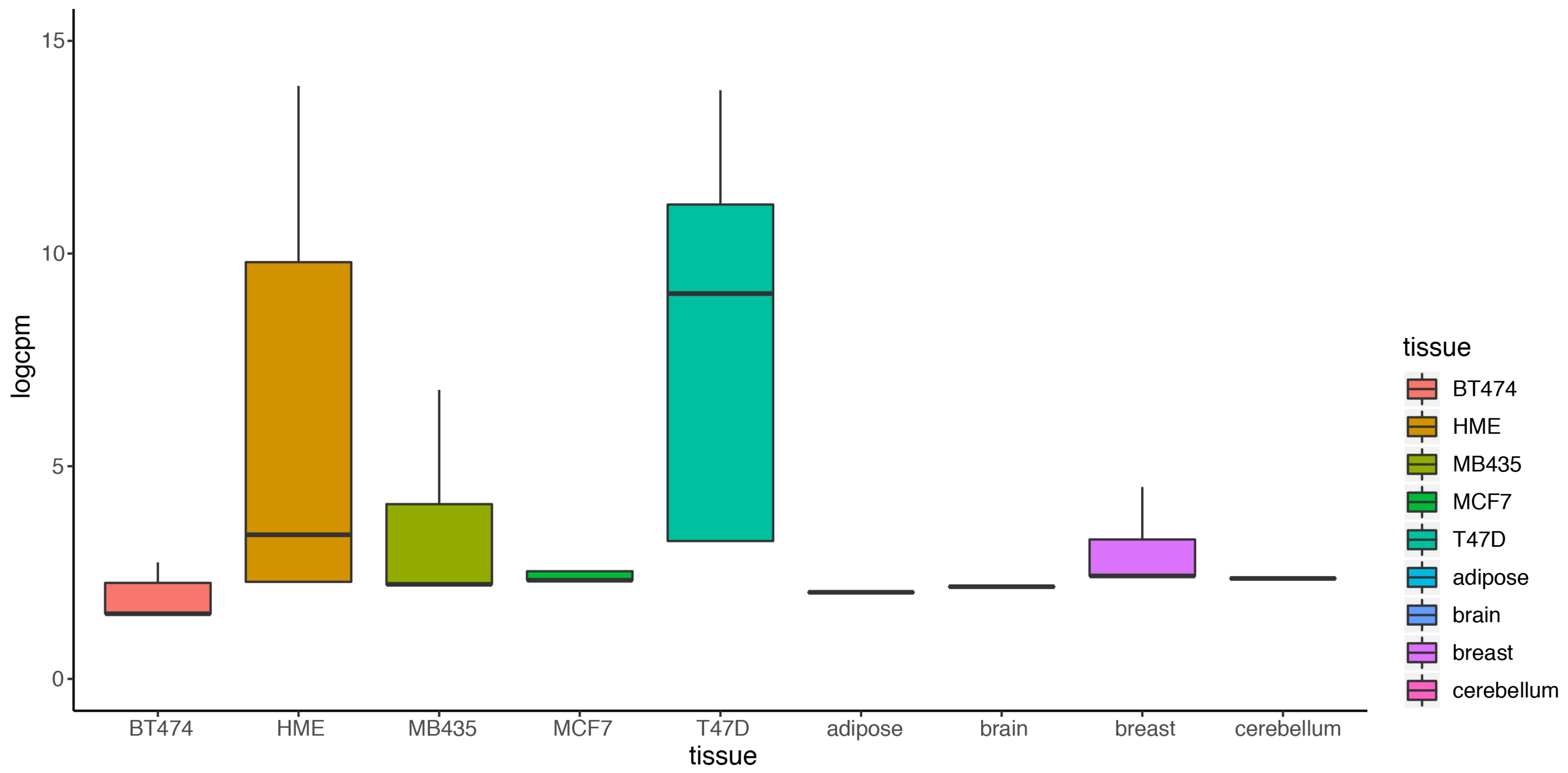
2.2. DEF Successfully Identified the Differentially Expressed Genes under the Real Dataset
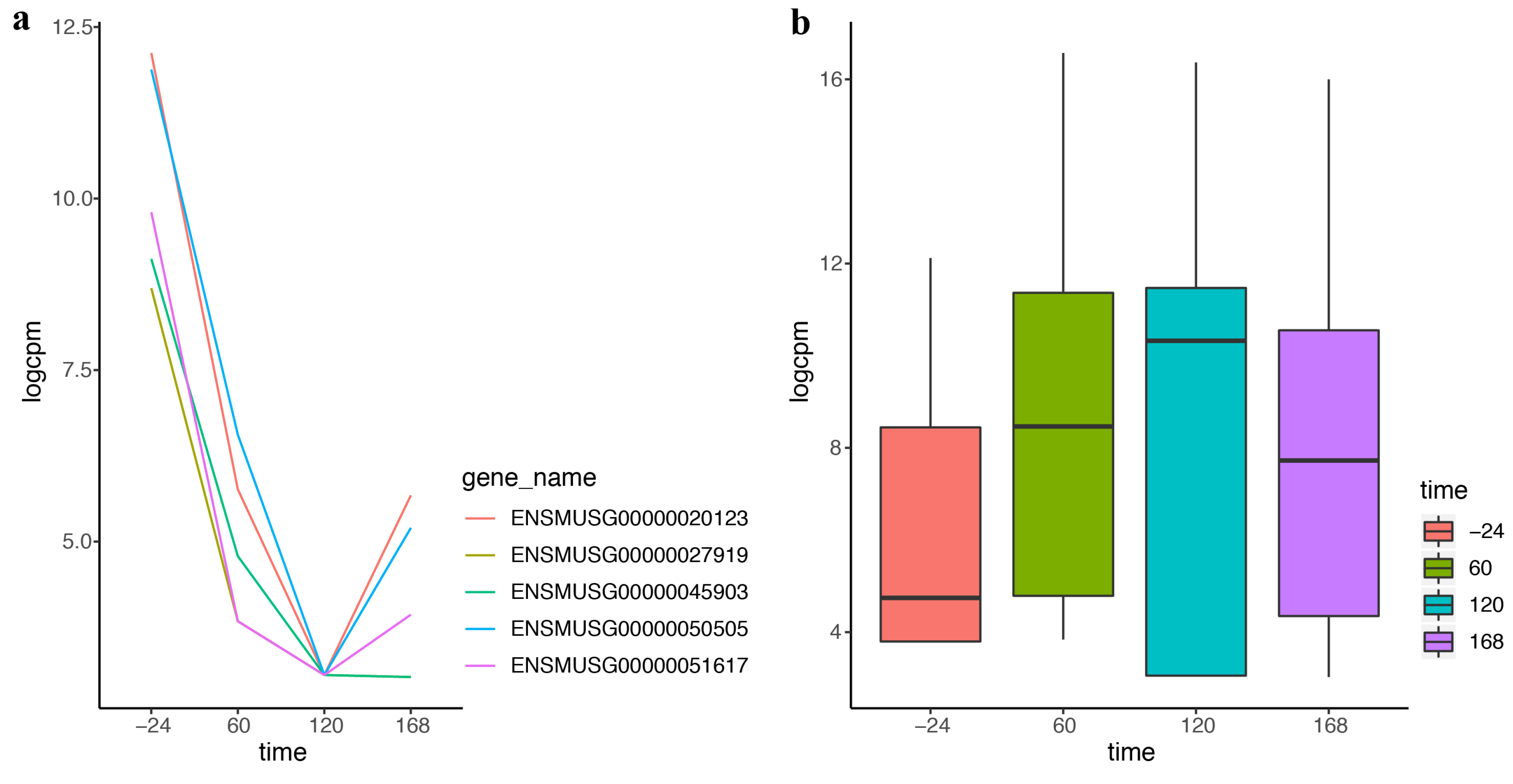
2.3. DEF Is Applicable for the Real Time-Course Data
2.4. DEF Is Applicable for the Real Multiple Condition Data
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Dataset
5.2. Normalization
5.3. DEF Function
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEF | Differential Entropy-like Function |
| NGS | Next Generation Sequencing |
| DEG | Differentially Expressed Gene |
References
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavijo, B.J.; Venturini, L.; Schudoma, C.; Accinelli, G.G.; Kaithakottil, G.; Wright, J.; Borrill, P.; Kettleborough, G.; Heavens, D.; Chapman, H.; et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017, 27, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chepelev, I.; Wei, G.; Tang, Q.; Zhao, K. Detection of single nucleotide variations in expressed exons of the human genome using RNA-Seq. Nucleic Acids Res. 2009, 37, e106. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial Analysis of Gene Expression. Science 1995, 270, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Kodzius, R.; Kojima, M.; Nishiyori, H.; Nakamura, M.; Fukuda, S.; Tagami, M.; Sasaki, D.; Imamura, K.; Kai, C.; Harbers, M.; et al. CAGE: Cap analysis of gene expression. Nat. Methods 2006, 3, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Little, S.C.; Tikhonov, M.; Gregor, T. Precise Developmental Gene Expression Arises from Globally Stochastic Transcriptional Activity. Cell 2013, 154, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Valerio, C. RNA-Seq and Human Complex Disease. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef]
- Robinson, M.D.; Smyth, G.K. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics 2007, 23, 2881–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. Differential Expression Analysis of Complex RNA-seq Experiments Using edgeR. In Statistical Analysis of Next Generation Sequencing Data; Datta, S., Nettleton, D., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 51–74. [Google Scholar] [CrossRef] [Green Version]
- Lun, A.T.L.; Chen, Y.; Smyth, G.K. It’s DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Springer: New York, NY, USA, 2016; pp. 391–416. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, T.J.; Kelly, K.A. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinform. 2010, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Witten, D.M.; Johnstone, I.M.; Tibshirani, R. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics 2012, 13, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.M.; Fisher, A.P.; Sozzani, R. Identifying Differentially Expressed Genes Using Fluorescence-Activated Cell Sorting (FACS) and RNA Sequencing from Low Input Samples. In Computational Cell Biology: Methods and Protocols; von Stechow, L., Santos Delgado, A., Eds.; Springer: New York, NY, USA, 2018; pp. 139–151. [Google Scholar] [CrossRef]
- Wu, H.; Wang, C.; Wu, Z. A new shrinkage estimator for dispersion improves differential expression detection in RNA-seq data. Biostatistics 2013, 14, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Vavoulis, D.V.; Francescatto, M.; Heutink, P.; Gough, J. DGEclust: Differential expression analysis of clustered count data. Genome Biol. 2015, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Golovnina, K.; Chen, Z.X.; Lee, H.N.; Negron, Y.L.S.; Sultana, H.; Oliver, B.; Harbison, S.T. Comparison of normalization and differential expression analyses using RNA-Seq data from 726 individual Drosophila melanogaster. BMC Genom. 2016, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Chinnaiyan, A.M.; Jiang, H. rSeqNP: A non-parametric approach for detecting differential expression and splicing from RNA-Seq data. Bioinformatics 2015, 31, 2222–2224. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tibshirani, R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-Seq data. Stat. Methods Med. Res. 2013, 22, 519–536. [Google Scholar] [CrossRef] [PubMed]
- Frazee, A.C.; Langmead, B.; Leek, J.T. ReCount: A multi-experiment resource of analysis-ready RNA-seq gene count datasets. BMC Bioinform. 2011, 12, 449. [Google Scholar] [CrossRef] [PubMed]
- Rivkin, E.; Eddy, E.M.; Willis, W.D.; Goulding, E.H.; Suganuma, R.; Yanagimachi, R.; Kierszenbaum, A.L. Sperm tail abnormalities in mutant mice with neor gene insertion into an intron of the keratin 9 gene. Mol. Reprod. Dev. 2005, 72, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.Y.; Klar, J.; Georgii-Heming, P.; Fröjmark, A.S.; Baig, S.M.; Schlessinger, D.; Dahl, N. Frizzled6 Deficiency Disrupts the Differentiation Process of Nail Development. J. Investig. Dermatol. 2013, 133, 1990–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, V.G.; Nayak, R.R.; Wang, I.X.; Elwyn, S.; Cousins, S.M.; Morley, M.; Spielman, R.S. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol. 2010, 8, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Plant, K.; Fairfax, B.P.; Makino, S.; Vandiedonck, C.; Radhakrishnan, J.; Knight, J.C. Fine mapping genetic determinants of the highly variably expressed MHC gene ZFP57. Eur. J. Hum. Genet. 2013, 22, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, J.; Künstle, G.; Obata, T.; Sha, M.; Noguchi, M. The Protooncogene TCL1 Is an Akt Kinase Coactivator. Mol. Cell 2000, 6, 395–407. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Koval, A.; Hallas, C.; Bichi, R.; Tresini, M.; Malstrom, S.; Russo, G.; Tsichlis, P.; Croce, C.M. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 3028–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherle, P.; Behrens, T.; Staudt, L. Ly-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proc. Natl. Acad. Sci. USA 1993, 90, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Adra, C.; Ko, J.; Leonard, D.; Wirth, L.; Cerione, R.; Lim, B. Identification of a novel protein with GDP dissociation inhibitor activity for the ras-like proteins CDC42Hs and rac I. Genes Chromosomes Cancer 1993, 8, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Leffers, H.; Nielsen, M.; Andersen, A.; Honoré, B.; Madsen, P.; Vandekerckhove, J.; Celis, J. Identification of two human Rho GDP dissociation inhibitor proteins whose overexpression leads to disruption of the actin cytoskeleton. Exp. Cell Res. 1993, 209, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Fantin, V.; Sparling, J.; Slot, J.; Keller, S.; Lienhard, G.; Lavan, B. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J. Biol. Chem. 1998, 273, 10726–10732. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Karas, M.; Koval, A.; LeRoith, D. Insulin receptor substrate-4 enhances insulin-like growth factor-I-induced cell proliferation. J. Biol. Chem. 1999, 274, 31179–31184. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, E.P.; Escribano, O.; Chiloeches, A.; Ramirez Rubio, S.; Román, I.D.; Fernández-Moreno, M.D.; Guijarro, L.G. Role of insulin receptor substrate-4 in IGF-I-stimulated HEPG2 proliferation. J. Hepatol. 2007, 46, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Marc, S.; Schulz, M.H.; Hugues, R.; Alon, M.; Andreas, K.; Matthias, S.; Martin, S.; Tatjana, B.; Aleksey, S.; Dmitri, P. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 2008, 321, 956–960. [Google Scholar]
- Katz, Y.; Wang, E.T.; Airoldi, E.M.; Burge, C.B. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat. Methods 2010, 7, 1009–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Rickard, S.; Shujun, L.; Irina, K.; Lu, Z.; Christine, M.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ensembl ID | Condition A | Replicate 2 | Condition B | Replicate 2 | DEF Value |
|---|---|---|---|---|---|
| Replicate 1 | Replicate 1 | ||||
| ENSG00000164002 | 5 | 0 | 0 | 0 | 0.0617 |
| ENSG00000104833 | 0 | 0 | 7 | 0 | 0.0608 |
| ENSG00000124920 | 0 | 0 | 7 | 0 | 0.0608 |
| ENSG00000182310 | 0 | 0 | 7 | 0 | 0.0608 |
| ENSG00000197608 | 0 | 0 | 0 | 6 | 0.0581 |
| Ensembl ID | Condition A | Replicate 2 | Condition B | Replicate 2 | DEF Value |
|---|---|---|---|---|---|
| Replicate 1 | Replicate 1 | ||||
| ENSG00000111325 | 4 | 8 | 6 | 2 | 0.0101 |
| ENSG00000141431 | 1 | 1 | 5 | 4 | 0.0100 |
| ENSG00000065357 | 4 | 10 | 3 | 4 | 0.0100 |
| ENSG00000179021 | 2 | 6 | 6 | 2 | 0.0100 |
| ENSG00000215301 | 2 | 6 | 6 | 2 | 00100 |
| Ensembl ID | Condition A | Replicate 2 | Condition B | Replicate 2 | DEF Value |
|---|---|---|---|---|---|
| Replicate 1 | Replicate 1 | ||||
| ENSMUSG00000051920 | 0 | 0 | 5 | 0 | 0.0437 |
| ENSMUSG00000029683 | 0 | 0 | 0 | 4 | 0.0436 |
| ENSMUSG00000069301 | 0 | 0 | 0 | 4 | 0.0436 |
| ENSMUSG00000070691 | 0 | 0 | 1 | 4 | 0.0432 |
| ENSMUSG00000079332 | 0 | 0 | 3 | 2 | 0.0412 |
| Ensembl ID | Condition A | Replicate 2 | Condition B | Replicate 2 | DEF Value |
|---|---|---|---|---|---|
| Replicate 1 | Replicate 1 | ||||
| ENSMUSG00000038593 | 6 | 3 | 4 | 0 | 0.0101 |
| ENSMUSG00000036977 | 5 | 11 | 0 | 1 | 0.0101 |
| ENSMUSG00000057924 | 2 | 2 | 5 | 1 | 0.0101 |
| ENSMUSG00000067203 | 2 | 2 | 5 | 1 | 0.0101 |
| ENSMUSG00000002205 | 2 | 13 | 10 | 3 | 0.0100 |
| Ensembl ID | Gene Symbol | Gene Function |
|---|---|---|
| ENSMUSG00000051617 | Krt9 | An important special function in the mature palmar |
| Plays an essential role in the correct development of sperm | ||
| ENSMUSG00000020123 | Avpr1a | Receptor for arginine vasopressin |
| ENSMUSG00000050505 | Pcdh20 | Potential calcium-dependent cell-adhesion protein. |
| ENSMUSG00000027919 | Lce1g | keratinocyte differentiation |
| ENSMUSG00000045903 | Npas4 | A key role in the structural and functional plasticity of neurons |
| Transcription factor expressed in neurons of the brain |
| Ensembl ID | Gene Symbol | Gene Function |
|---|---|---|
| ENSG00000204644 | ZFP57 | May serve an important special function either in the mature palmar |
| Plays an essential role in the correct development of sperm | ||
| ENSG00000007038 | PRSS21 | Receptor for arginine vasopressin |
| ENSG00000179455 | MKRN | Potential calcium-dependent cell-adhesion protein. |
| ENSG00000170627 | GTSF1 | Protein coding |
| ENSG00000129824 | RPS4Y1 | multicellular organism development |
| nuclear-transcribed mRNA catabolic process, nonsense-mediated decay |
| Ensembl ID | Gene Symbol | Gene Function |
|---|---|---|
| ENSG00000100721 | TCL1A | Enhances cell proliferation, stabilizes mitochondrial membrane potential and promotes cell survival |
| Enhances the phosphorylation and activation of AKT1, AKT2 and AKT3. | ||
| ENSG00000110777 | POU2AF1 | It is essential for the response of B-cells to antigens and required for the formation of germinal centers |
| ENSG00000111348 | ARHGDIB | Regulates the GDP/GTP exchange reaction of the Rho proteins by inhibiting the dissociation of GDP from them, and the subsequent binding of GTP to them |
| ENSG00000118308 | LRMP | Plays a role in the delivery of peptides to major histocompatibility complex (MHC) class I molecules |
| May play a role during fertilization in pronucleus congression and fusion | ||
| ENSG00000133124 | IRS4 | Acts as an interface between multiple growth factor receptors possessing tyrosine kinase activity |
| Plays a pivotal role in the proliferation/differentiation of hepatoblastoma cell | ||
| Plays a role in growth, reproduction and glucose homeostasis |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Jin, S.; Zhang, C. A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies. Entropy 2019, 21, 242. https://doi.org/10.3390/e21030242
Wang Z, Jin S, Zhang C. A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies. Entropy. 2019; 21(3):242. https://doi.org/10.3390/e21030242
Chicago/Turabian StyleWang, Zhuo, Shuilin Jin, and Chiping Zhang. 2019. "A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies" Entropy 21, no. 3: 242. https://doi.org/10.3390/e21030242
APA StyleWang, Z., Jin, S., & Zhang, C. (2019). A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies. Entropy, 21(3), 242. https://doi.org/10.3390/e21030242