Role of Entropy in Colloidal Self-Assembly



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Entropic Contributions in Self-Assembly Processes
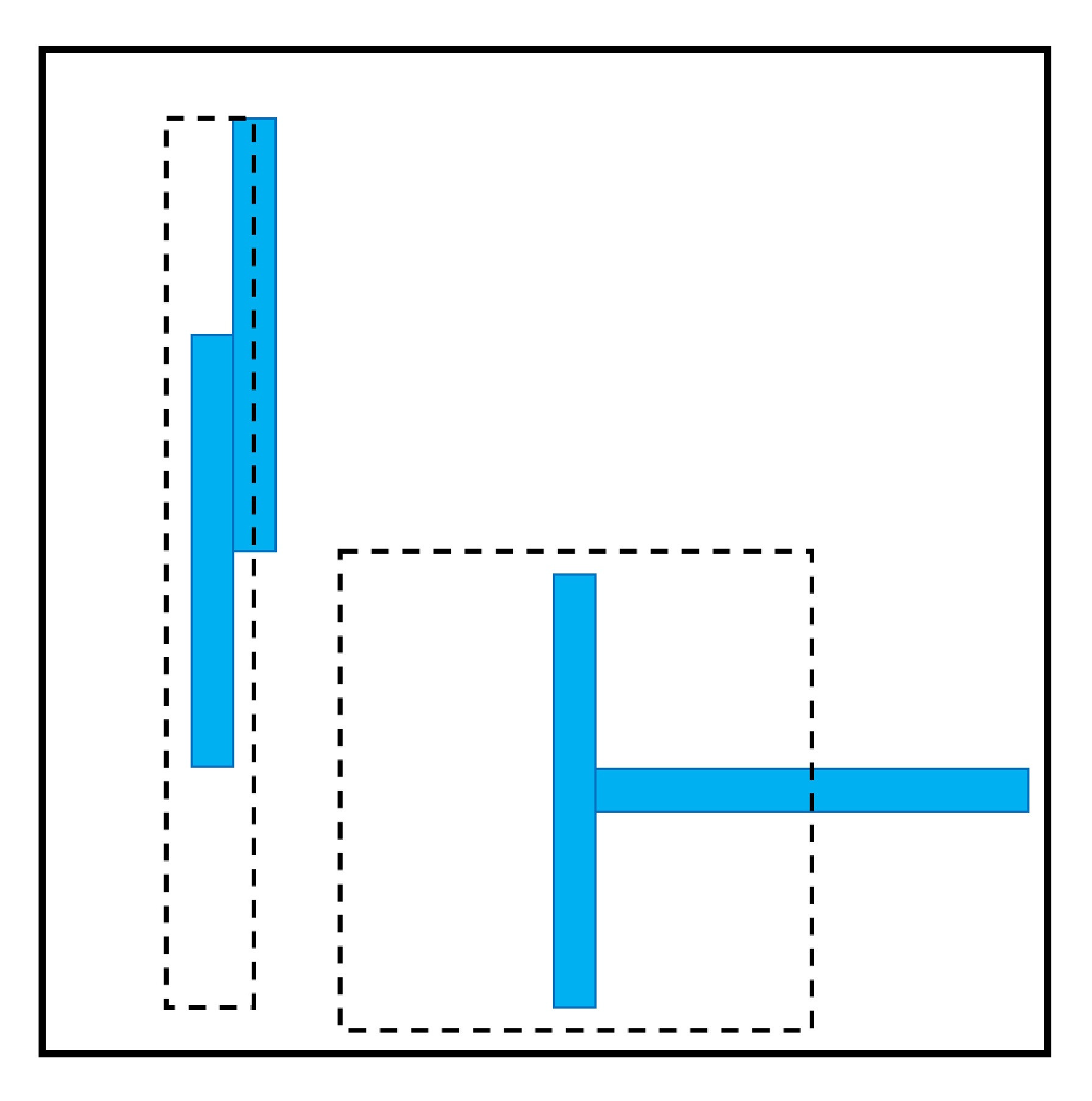
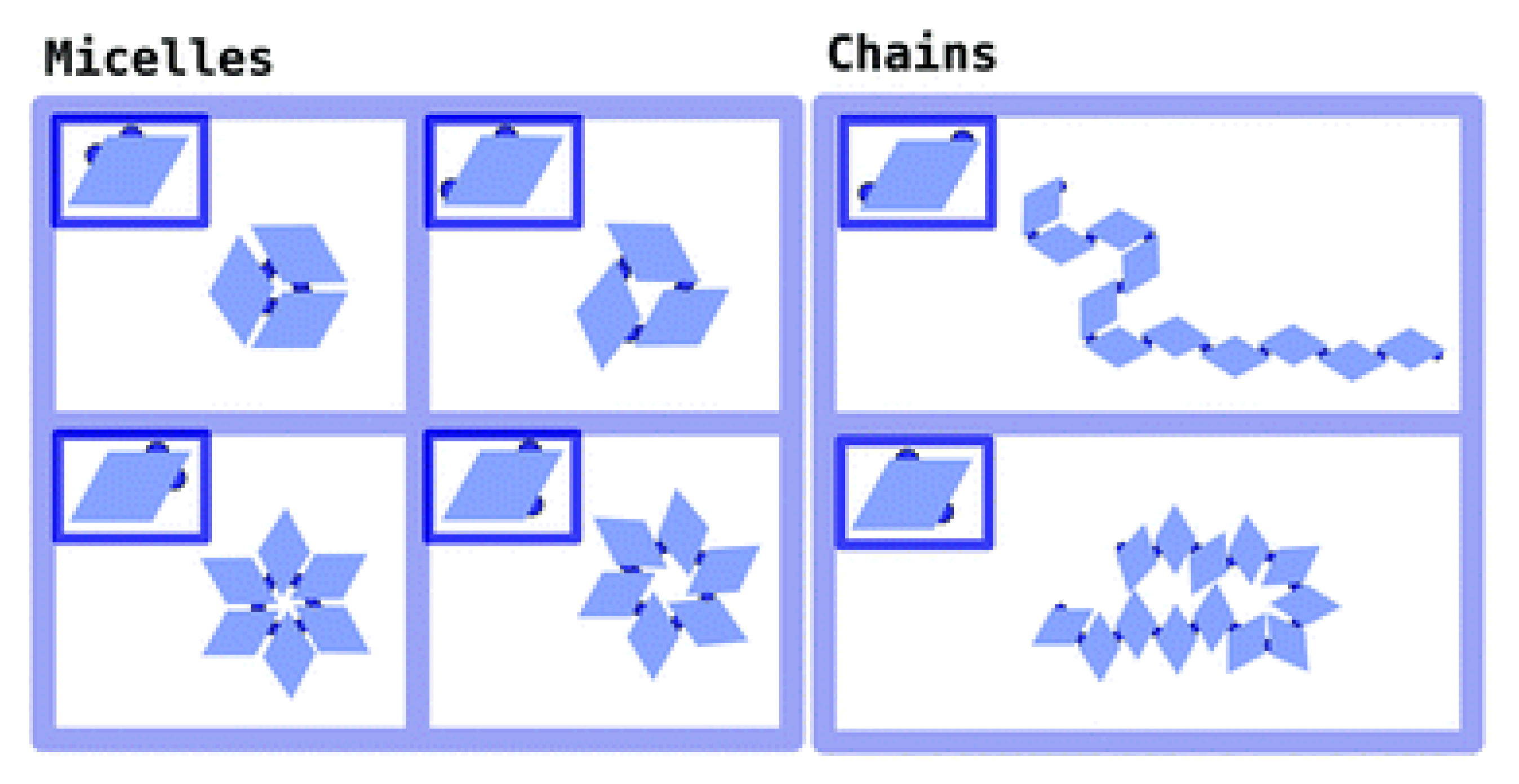
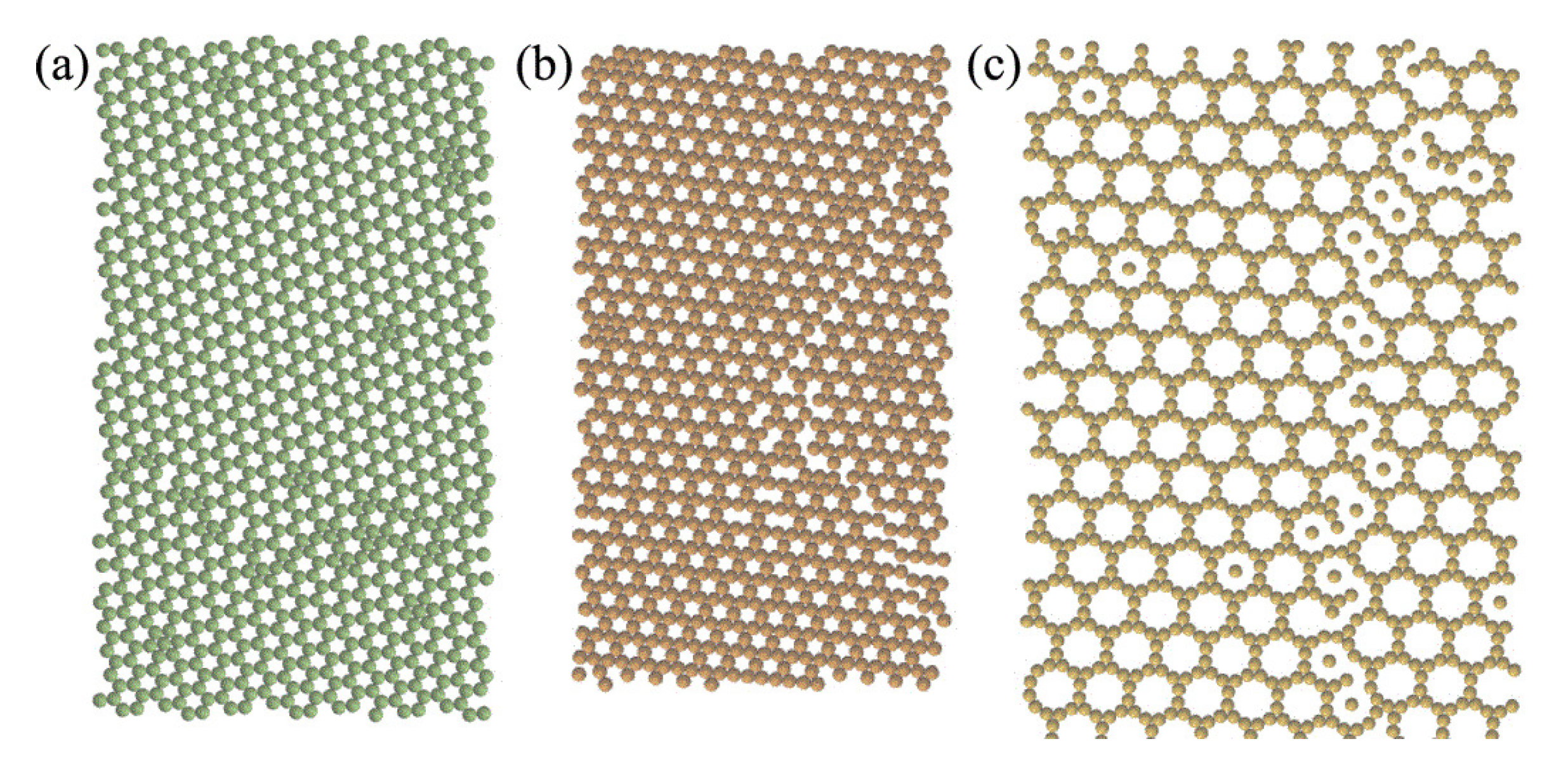
2.1. Effect of Particle Shape in Self-Assembly
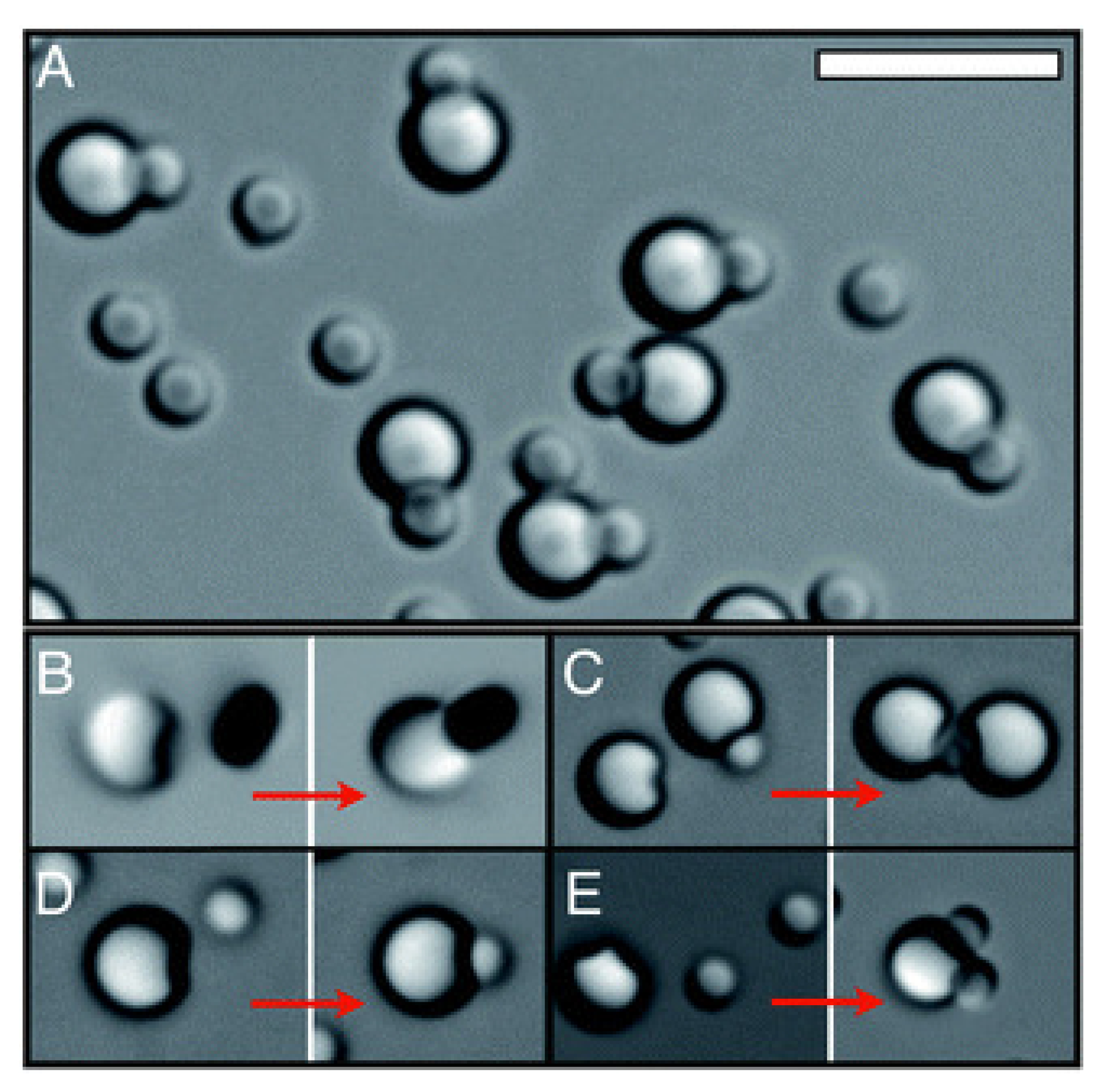
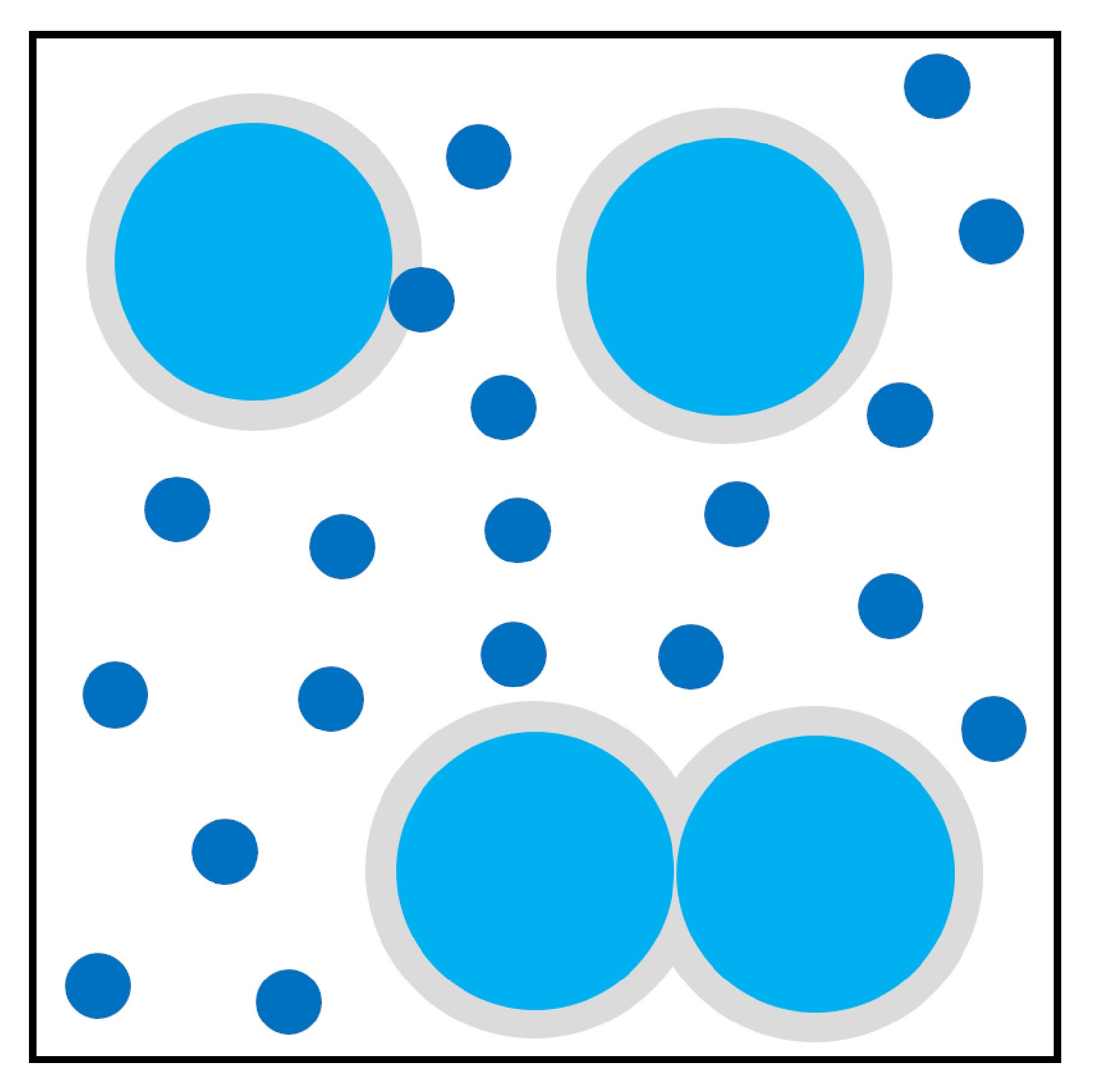
2.2. Self-Assembly Driven by Depletion Effects
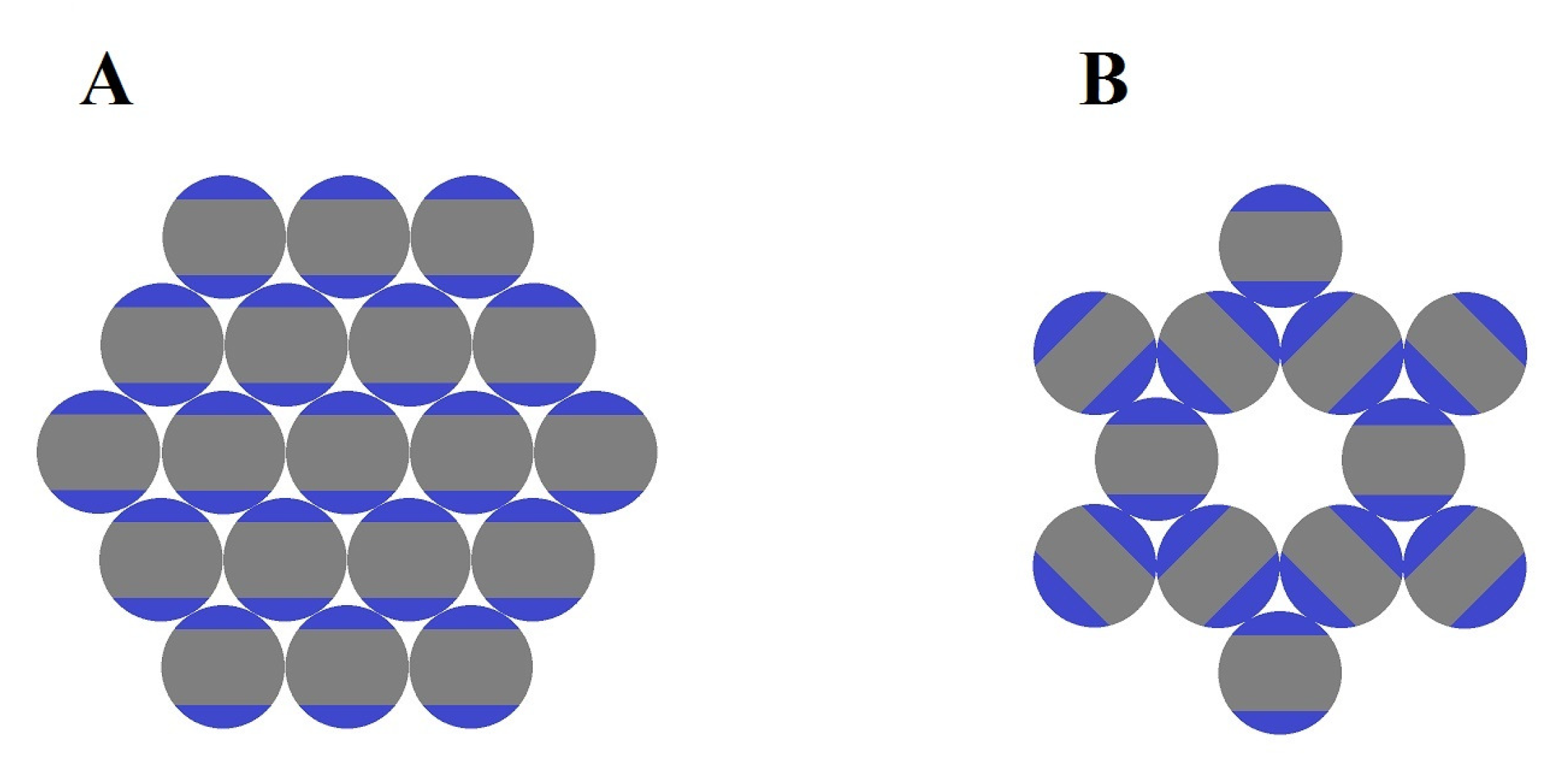
2.3. Role of Entropy in the Formation of Open Lattices
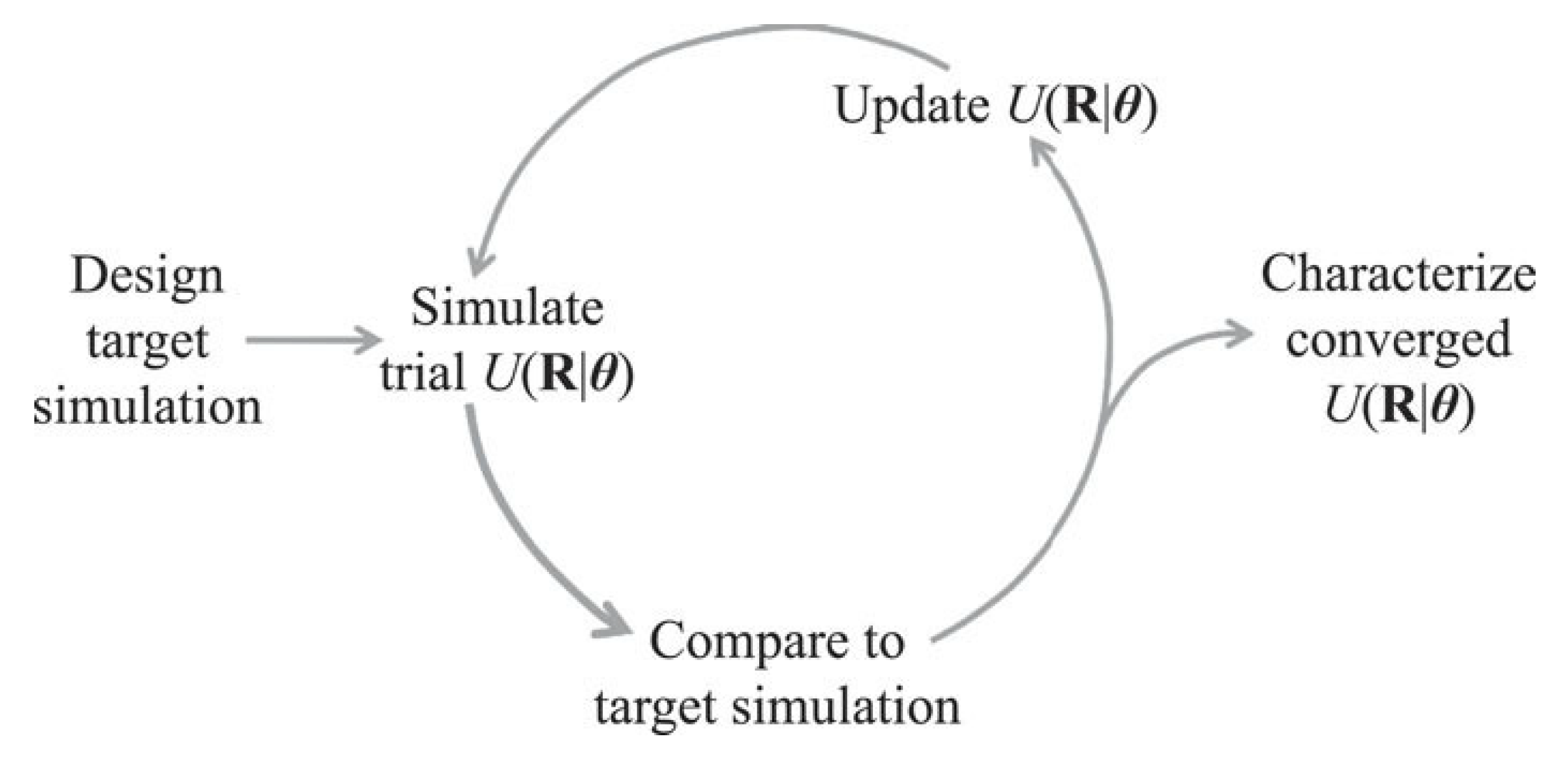
2.4. Relative Entropy and Its Importance in Inverse Design Techniques
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MC | Monte Carlo |
| MD | Molecular Dynamics |
| CG | Coarse-Grained |
References
- Clausius, R. Ueber verschiedene für die anwendung bequeme formen der hauptgleichungen der mechanischen wärmetheorie. Ann. Phys. 1865, 125, 353–400. [Google Scholar] [CrossRef] [Green Version]
- Clausius, R.; Hirst, T.; Tyndall, J. The Mechanical Theory of Heat: With Its Applications to the Steam-Engine and to the Physical Properties of Bodies; John Van Voorst: London, UK, 1867. [Google Scholar]
- Callen, H. Thermodynamics and an Introduction to Thermostatics; John Wiley & Sons, Inc.: New York, NY, USA, 1985. [Google Scholar]
- Hill, T.L. An Introduction to Statistical Thermodynamics; Dover Publications: New York, NY, USA, 1986. [Google Scholar]
- Shell, M.S. Thermodynamics and Statistical Mechanics: An Integrated Approach; Cambridge Series in Chemical Engineering; Cambridge University Press: Cambridge, UK, 2015. [Google Scholar]
- Planck, M. Ueber das Gesetz der Energieuerteilzcng im Norrnalspectrzcm. Ann. Phys. 1901, 309, 553–563. [Google Scholar] [CrossRef]
- Planck, M. Treatise on Thermodynamics; Dover Publications: New York, NY, USA, 2010. [Google Scholar]
- Mammen, M.; Shakhnovich, E.I.; Deutch, J.M.; Whitesides, G.M. Estimating the entropic cost of self-assembly of multiparticle hydrogen-bonded aggregates based on the cyanuric acidmelamine lattice. J. Org. Chem. 1998, 63, 3821–3830. [Google Scholar] [CrossRef]
- Harano, Y.; Kinoshita, M. Crucial importance of translational entropy of water in pressure denaturation of proteins. J. Chem. Phys. 2006, 125, 024910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, M.A.; Engel, M.; Talapin, D.V. Self-assembly of colloidal nanocrystals: From intricate structures to functional materials. Chem. Rev. 2016, 116, 11220–11289. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Boncheva, M. Beyond molecules: Self-assembly of mesoscopic and macroscopic components. Proc. Natl. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar] [CrossRef] [Green Version]
- Grzelczak, M.; Vermant, J.; Furst, E.M.; Liz-Marzán, L.M. Directed self-assembly of nanoparticles. ACS Nano 2010, 4, 3591–3605. [Google Scholar] [CrossRef]
- Pretti, E.; Zerze, H.; Song, M.; Ding, Y.; Mahynski, N.A.; Hatch, H.W.; Shen, V.K.; Mittal, J. Assembly of three-dimensional binary superlattices from multi-flavored particles. Soft Matter 2018, 14, 6303–6312. [Google Scholar] [CrossRef]
- Song, M.; Ding, Y.; Zerze, H.; Snyder, M.A.; Mittal, J. Binary superlattice design by controlling DNA-mediated interactions. Langmuir 2018, 34, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.A.; Kamita, G.; Kurokawa, T.; Tsujii, K.; Gong, J.P. Unidirectional alignment of lamellar bilayer in hydrogel: One-dimensional swelling, anisotropic modulus, and stress/strain tunable structural color. Adv. Mater 2010, 22, 5110–5114. [Google Scholar] [CrossRef]
- Singh, G.; Chan, H.; Baskin, A.; Gelman, E.; Repnin, N.; Král, P.; Klajn, R. Self-assembly of magnetite nanocubes into helical superstructures. Science 2014, 345, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Rogers, W.B.; Shih, W.M.; Manoharan, V.N. Using DNA to program the self-assembly of colloidal nanoparticles and microparticles. Nat. Rev. Mater. 2016, 1, 16008. [Google Scholar] [CrossRef]
- Frenkel, D. Entropy-driven phase transitions. Physica A 1999, 263, 26–38. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, C.K.; Hamblin, G.D.; Sleiman, H.F. Supramolecular DNA assembly. Chem. Soc. Rev. 2011, 40, 5647. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Yang, S.; Yan, Y.; Huang, J.; Petukhov, A.V.; Kroon-Batenburg, L.M.J.; Drechsler, M.; Zhou, C.; Tu, M.; Granick, S.; Jiang, L. Giant capsids from lattice self-assembly of cyclodextrin complexes. Nat. Commun. 2017, 8, 15856. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Petukhova, A.; Kumacheva, E. Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotechnol. 2009, 5, 15–25. [Google Scholar]
- Zhang, J.; Li, Y.; Zhang, X.; Yang, B. Colloidal self-assembly meets nanofabrication: From two-dimensional colloidal crystals to nanostructure arrays. Adv. Mater. 2010, 22, 4249–4269. [Google Scholar] [CrossRef]
- Lu, P.J.; Weitz, D.A. Colloidal particles: Crystals, glasses, and gels. Annu. Rev. Condens. Matter Phys. 2013, 4, 217–233. [Google Scholar]
- Frenkel, D. Order through entropy. Nat. Mater. 2014, 14, 9–12. [Google Scholar] [CrossRef]
- Harper, E.S.; van Anders, G.; Glotzer, S.C. The entropic bond in colloidal crystals. Proc. Natl. Acad. Sci. USA 2019, 116, 16703–16710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swendsen, R.H. Gibbs’ paradox and the definition of entropy. Entropy 2008, 10, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Frenkel, D. Why colloidal systems can be described by statistical mechanics: Some not very original comments on the Gibbs paradox. Mol. Phys. 2014, 112, 2325–2329. [Google Scholar] [CrossRef] [Green Version]
- Willard Gibbs, J. Elementary Principles of Statistical Mechanics; Yale University Press: New Haven, CT, USA, 1902. [Google Scholar]
- Onsager, L. The effects of shape on the interaction of colloidal particles. Ann. N. Y. Acad. Sci. 1949, 51, 627–659. [Google Scholar] [CrossRef]
- Vroege, G.J.; Lekkerkerker, H.N. Phase transitions in lyotropic colloidal and polymer liquid crystals. Rep. Prog. Phys. 1992, 55, 1241. [Google Scholar] [CrossRef]
- Wood, W.W.; Jacobson, J.D. Preliminary results from a recalculation of the Monte Carlo equation of state of hard spheres. J. Chem. Phys. 1957, 27, 1207–1208. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Phase transition for a hard sphere system. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef] [Green Version]
- Kofke, D.A.; Bolhuis, P.G. Freezing of polydisperse hard spheres. Phys. Rev. E 1999, 59, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Truskett, T.M.; Torquato, S.; Sastry, S.; Debenedetti, P.G.; Stillinger, F.H. Structural precursor to freezing in the hard-disk and hard-sphere systems. Phys. Rev. E 1998, 58, 3083–3088. [Google Scholar] [CrossRef] [Green Version]
- Asakura, S.; Oosawa, F. On interaction between two bodies immersed in a solution of macromolecules. J. Chem. Phys. 1954, 22, 1255–1256. [Google Scholar] [CrossRef]
- Evers, W.H.; Nijs, B.D.; Filion, L.; Castillo, S.; Dijkstra, M.; Vanmaekelbergh, D. Entropy-driven formation of binary semiconductor-nanocrystal superlattices. Nano Lett. 2010, 10, 4235–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nijs, B.; Dussi, S.; Smallenburg, F.; Meeldijk, J.D.; Groenendijk, D.J.; Filion, L.; Imhof, A.; van Blaaderen, A.; Dijkstra, M. Entropy-driven formation of large icosahedral colloidal clusters by spherical confinement. Nat. Mater. 2014, 14, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Bodnarchuk, M.I.; Kovalenko, M.V.; Heiss, W.; Talapin, D.V. Energetic and entropic contributions to self-assembly of binary nanocrystal superlattices: Temperature as the structure-directing factor. J. Am. Chem. Soc. 2010, 132, 11967–11977. [Google Scholar] [CrossRef] [PubMed]
- Storhoff, J.J.; Lazarides, A.A.; Mucic, R.C.; Mirkin, C.A.; Letsinger, R.L.; Schatz, G.C. What controls the optical properties of DNA-linked gold nanoparticle assemblies? J. Am. Chem. Soc. 2000, 122, 4640–4650. [Google Scholar] [CrossRef]
- Park, S.J.; Taton, T.A.; Mirkin, C.A. Array-based electrical detection of DNA with nanoparticle probes. Science 2002, 295, 1503–1506. [Google Scholar]
- Jin, R.; Wu, G.; Li, Z.; Mirkin, C.A.; Schatz, G.C. What controls the melting properties of DNA-linked gold nanoparticle assemblies? J. Am. Chem. Soc. 2003, 125, 1643–1654. [Google Scholar] [CrossRef]
- Soto, C.M.; Srinivasan, A.; Ratna, B.R. Controlled assembly of mesoscale structures using DNA as molecular bridges. J. Am. Chem. Soc. 2002, 124, 8508–8509. [Google Scholar] [CrossRef]
- Rogers, W.B.; Crocker, J.C. Direct measurements of DNA-mediated colloidal interactions and their quantitative modeling. Proc. Natl. Acad. Sci. USA 2011, 108, 15687–15692. [Google Scholar] [CrossRef] [Green Version]
- Mladek, B.M.; Fornleitner, J.; Martinez-Veracoechea, F.J.; Dawid, A.; Frenkel, D. Quantitative prediction of the phase diagram of DNA-functionalized nanosized colloids. Phys. Rev. Lett. 2012, 108, 268301. [Google Scholar] [CrossRef]
- Yue, Y.; Norikane, Y. Gold clay from self-assembly of 2D microscale nanosheets. Nat. Commun. 2020, 11, 568. [Google Scholar] [CrossRef]
- Lekkerkerker, H.N.W.; Stroobants, A. Ordering entropy. Nature 1998, 393, 305–307. [Google Scholar] [CrossRef]
- Packwood, D.M.; Han, P.; Hitosugi, T. Chemical and entropic control on the molecular self-assembly process. Nat. Commun. 2017, 8, 14463. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, C.; Escobedo, F.A. Packing, entropic patchiness, and self-assembly of non-convex colloidal particles: A simulation perspective. Curr. Opin. Colloid Interface Sci. 2017, 30, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Glotzer, S.C.; Solomon, M.J. Anisotropy of building blocks and their assembly into complex structures. Nat. Mater. 2007, 6, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hermes, M.; Kotni, R.; Wu, Y.; Tasios, N.; Liu, Y.; de Nijs, B.; van der Wee, E.B.; Murray, C.B.; Dijkstra, M.; et al. Interplay between spherical confinement and particle shape on the self-assembly of rounded cubes. Nat. Commun. 2018, 9, 2228. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, V.N. Colloidal matter: Packing, geometry, and entropy. Science 2015, 349, 1253751. [Google Scholar] [CrossRef] [Green Version]
- Coluzza, I.; van Oostrum, P.D.J.; Capone, B.; Reimhult, E.; Dellago, C. Sequence controlled self-knotting colloidal patchy polymers. Phys. Rev. Lett. 2013, 110, 075501. [Google Scholar] [CrossRef]
- Coluzza, I.; van Oostrum, P.D.J.; Capone, B.; Reimhult, E.; Dellago, C. Design and folding of colloidal patchy polymers. Soft Matter 2013, 9, 938–944. [Google Scholar] [CrossRef]
- Cademartiri, L.; Bishop, K.J.M.; Snyder, P.W.; Ozin, G.A. Using shape for self-assembly. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2012, 370, 2824–2847. [Google Scholar] [CrossRef]
- Sacanna, S.; Korpics, M.; Rodriguez, K.; Colón-Meléndez, L.; Kim, S.H.; Pine, D.J.; Yi, G.R. Shaping colloids for self-assembly. Nat. Commun. 2013, 4, 1688. [Google Scholar] [CrossRef]
- Sacanna, S.; Irvine, W.T.M.; Chaikin, P.M.; Pine, D.J. Lock and key colloids. Nature 2010, 464, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Sacanna, S.; Irvine, W.T.M.; Rossi, L.; Pine, D.J. Lock and key colloids through polymerization-induced buckling of monodisperse silicon oil droplets. Soft Matter 2011, 7, 1631–1634. [Google Scholar] [CrossRef]
- Miszta, K.; de Graaf, J.; Bertoni, G.; Dorfs, D.; Brescia, R.; Marras, S.; Ceseracciu, L.; Cingolani, R.; van Roij, R.; Dijkstra, M.; et al. Hierarchical self-assembly of suspended branched colloidal nanocrystals into superlattice structures. Nat. Mater. 2011, 10, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Loudet, J.C.; Alsayed, A.M.; Zhang, J.; Yodh, A.G. Capillary interactions between anisotropic colloidal particles. Phys. Rev. Lett. 2005, 94, 018301. [Google Scholar] [CrossRef] [PubMed]
- Akcora, P.; Liu, H.; Kumar, S.K.; Moll, J.; Li, Y.; Benicewicz, B.C.; Schadler, L.S.; Acehan, D.; Panagiotopoulos, A.Z.; Pryamitsyn, V.; et al. Anisotropic self-assembly of spherical polymer-grafted nanoparticles. Nat. Mater. 2009, 8, 354–359. [Google Scholar] [CrossRef]
- Ellison, L.J.; Michel, D.J.; Barmes, F.; Cleaver, D.J. Entropy-driven formation of the gyroid cubic phase. Phys. Rev. Lett. 2006, 97, 237801. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Vashisth, H. Self-assembly of lobed particles into amorphous and crystalline porous structures. Soft Matter 2020, 16, 1142–1147. [Google Scholar] [CrossRef]
- Karner, C.; Dellago, C.; Bianchi, E. How patchiness controls the properties of chain-like assemblies of colloidal platelets. J. Phys. Condens. Matter 2020, 32, 204001. [Google Scholar] [CrossRef] [Green Version]
- Karner, C.; Dellago, C.; Bianchi, E. Hierarchical self-assembly of patchy colloidal platelets. Soft Matter 2020, 16, 2774–2785. [Google Scholar] [CrossRef]
- Davis, J.R.; Panagiotopoulos, A.Z. Monte Carlo simulations of amphiphilic nanoparticle self-assembly. J. Chem. Phys. 2008, 129, 194706. [Google Scholar] [CrossRef]
- Damasceno, P.F.; Engel, M.; Glotzer, S.C. Crystalline assemblies and densest packings of a family of truncated tetrahedra and the role of directional entropic forces. ACS Nano 2012, 6, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Anders, G.; Klotsa, D.; Ahmed, N.K.; Engel, M.; Glotzer, S.C. Understanding shape entropy through local dense packing. Proc. Natl. Acad. Sci. USA 2014, 111, E4812–E4821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Anders, G.; Ahmed, N.K.; Smith, R.; Engel, M.; Glotzer, S.C. Entropically patchy particles: Engineering valence through shape entropy. ACS Nano 2014, 8, 931–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallenburg, F.; Filion, L.; Marechal, M.; Dijkstra, M. Vacancy-stabilized crystalline order in hard cubes. Proc. Natl. Acad. Sci. USA 2012, 109, 17886–17890. [Google Scholar] [CrossRef] [Green Version]
- Gantapara, A.P.; de Graaf, J.; van Roij, R.; Dijkstra, M. Phase diagram and structural diversity of a family of truncated cubes: Degenerate close-packed structures and vacancy-rich states. Phys. Rev. Lett. 2013, 111, 015501. [Google Scholar] [CrossRef]
- Asakura, S.; Oosawa, F. Interaction between particles suspended in solutions of macromolecules. J. Polym. Sci. 1958, 33, 183–192. [Google Scholar] [CrossRef]
- Dijkstra, M.; Frenkel, D. Evidence for entropy-driven demixing in hard-core fluids. Phy. Rev. Lett. 1994, 72, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Lubensky, T. Soft condensed matter physics. Solid State Commun. 1997, 102, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Yodh, A.G.; Lin, K.; Crocker, J.C.; Dinsmore, A.D.; Verma, R.; Kaplan, P.D. Entropically driven self–assembly and interaction in suspension. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2001, 359, 921–937. [Google Scholar] [CrossRef]
- Kraft, D.J.; Ni, R.; Smallenburg, F.; Hermes, M.; Yoon, K.; Weitz, D.A.; van Blaaderen, A.; Groenewold, J.; Dijkstra, M.; Kegel, W.K. Surface roughness directed self-assembly of patchy particles into colloidal micelles. Proc. Natl. Acad. Sci. USA 2012, 109, 10787–10792. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.J.M.; Wilmer, C.E.; Soh, S.; Grzybowski, B.A. Nanoscale forces and their uses in self-assembly. Small 2009, 5, 1600–1630. [Google Scholar] [CrossRef] [PubMed]
- Escobedo, F.A. Engineering entropy in soft matter: The bad, the ugly and the good. Soft Matter 2014, 10, 8388–8400. [Google Scholar] [CrossRef] [PubMed]
- Baranov, D.; Fiore, A.; van Huis, M.; Giannini, C.; Falqui, A.; Lafont, U.; Zandbergen, H.; Zanella, M.; Cingolani, R.; Manna, L. Assembly of colloidal semiconductor nanorods in solution by depletion attraction. Nano Lett. 2010, 10, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Zhang, Q.; Emrick, T.; Balazs, A.C.; Russell, T.P. Entropy-driven segregation of nanoparticles to cracks in multilayered composite polymer structures. Nat. Mater. 2006, 5, 229–233. [Google Scholar] [CrossRef]
- Zanella, M.; Bertoni, G.; Franchini, I.R.; Brescia, R.; Baranov, D.; Manna, L. Assembly of shape-controlled nanocrystals by depletion attraction. Chem. Commun. 2011, 47, 203–205. [Google Scholar] [CrossRef]
- Barry, E.; Dogic, Z. Entropy driven self-assembly of nonamphiphilic colloidal membranes. Proc. Natl. Acad. Sci. USA 2010, 107, 10348–10353. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Li, Y.; Wei, G.; Liu, Q.; Mundoor, H.; Chen, Z.; Smalyukh, I.I. Liquid crystal self-assembly of upconversion nanorods enriched by depletion forces for mesostructured material preparation. Nanoscale 2018, 10, 4218–4227. [Google Scholar] [CrossRef] [Green Version]
- Dinsmore, A.D.; Yodh, A.G. Entropic confinement of colloidal spheres in corners on silicon substrates. Langmuir 1999, 15, 314–316. [Google Scholar] [CrossRef]
- Kamp, M.; Hermes, M.; van Kats, C.M.; Kraft, D.J.; Kegel, W.K.; Dijkstra, M.; van Blaaderen, A. Selective depletion interactions in mixtures of rough and smooth silica spheres. Langmuir 2016, 32, 1233–1240. [Google Scholar] [CrossRef]
- Cates, M.E. Entropy stabilizes open crystals. Nat. Mater. 2013, 12, 179–180. [Google Scholar] [CrossRef]
- Mao, X. Entropic effects in the self-assembly of open lattices from patchy particles. Phys. Rev. E 2013, 87, 062319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Bae, S.C.; Granick, S. Directed self-assembly of a colloidal kagome lattice. Nature 2011, 469, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Chen, Q.; Granick, S. Entropy favours open colloidal lattices. Nat. Mater. 2013, 12, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Schulz, S.A.; Upham, J.; O’Faolain, L.; Boyd, R.W. Photonic crystal slow light waveguides in a kagome lattice. Opt. Lett. 2017, 42, 3243. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, A.; Long, C.; Choquette, K. Photonic crystal heterostructure cavity lasers using kagome lattices. Electron. Lett. 2008, 44, 803. [Google Scholar] [CrossRef] [Green Version]
- Gajić, R.; Meisels, R.; Kuchar, F.; Hingerl, K. All-angle left-handed negative refraction in Kagomé and honeycomb lattice photonic crystals. Phys. Rev. B 2006, 73, 165310. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, J.L.; Barrett, A.R.; Das, M.; Petersen, P.B.; Bonassar, L.J.; Cohen, I. Structure-function relations and rigidity percolation in the shear properties of articular cartilage. Biophys. J. 2014, 107, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Ruiz, P.S.; Ni, R. Entropy stabilizes floppy crystals of mobile DNA-coated colloids. Phys. Rev. Lett. 2018, 120, 048003. [Google Scholar] [CrossRef] [Green Version]
- Breen, T.L. Design and self-assembly of open, regular, 3D mesostructures. Science 1999, 284, 948–951. [Google Scholar] [CrossRef] [Green Version]
- Alberstein, R.; Suzuki, Y.; Paesani, F.; Tezcan, F.A. Engineering the entropy-driven free-energy landscape of a dynamic nanoporous protein assembly. Nat. Chem. 2018, 10, 732–739. [Google Scholar] [CrossRef]
- Shell, M.S. Coarse-graining with the relative entropy. In Advances in Chemical Physics; John Wiley & Sons, Inc.: New York, NY, USA, 2016; pp. 395–441. [Google Scholar]
- Chaimovich, A.; Shell, M.S. Relative entropy as a universal metric for multiscale errors. Phys. Rev. E 2010, 81, 060104. [Google Scholar] [CrossRef] [PubMed]
- Chaimovich, A.; Shell, M.S. Anomalous waterlike behavior in spherically-symmetric water models optimized with the relative entropy. Phys. Chem. Chem. Phys. 2009, 11, 1901. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, T.; Shell, M.S. Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation. J. Chem. Phys. 2016, 145, 034109. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.P.; Shell, M.S. A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J. Phys. Chem. B 2012, 116, 8383–8393. [Google Scholar] [CrossRef]
- Rechtsman, M.C.; Stillinger, F.H.; Torquato, S. Optimized interactions for targeted self-assembly: Application to a honeycomb lattice. Phys. Rev. Lett. 2005, 95, 228301. [Google Scholar] [CrossRef] [Green Version]
- Mahynski, N.A.; Mao, R.; Pretti, E.; Shen, V.K.; Mittal, J. Grand canonical inverse design of multicomponent colloidal crystals. Soft Matter 2020, 16, 3187–3194. [Google Scholar] [CrossRef]
- Adorf, C.S.; Antonaglia, J.; Dshemuchadse, J.; Glotzer, S.C. Inverse design of simple pair potentials for the self-assembly of complex structures. J. Chem. Phys. 2018, 149, 204102. [Google Scholar] [CrossRef] [Green Version]
- Van Anders, G.; Klotsa, D.; Karas, A.S.; Dodd, P.M.; Glotzer, S.C. Digital alchemy for materials design: Colloids and beyond. ACS Nano 2015, 9, 9542–9553. [Google Scholar] [CrossRef] [Green Version]
- Jadrich, R.B.; Lindquist, B.A.; Truskett, T.M. Probabilistic inverse design for self-assembling materials. J. Chem. Phys. 2017, 146, 184103. [Google Scholar] [CrossRef]
- Shell, M.S. The relative entropy is fundamental to multiscale and inverse thermodynamic problems. J. Chem. Phys. 2008, 129, 144108. [Google Scholar] [CrossRef]
- Piñeros, W.D.; Lindquist, B.A.; Jadrich, R.B.; Truskett, T.M. Inverse design of multicomponent assemblies. J. Chem. Phys. 2018, 148, 104509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Errington, J.R.; Truskett, T.M. Communication: Phase behavior of materials with isotropic interactions designed by inverse strategies to favor diamond and simple cubic lattice ground states. J. Chem. Phys. 2013, 139, 141102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindquist, B.A.; Jadrich, R.B.; Truskett, T.M. Communication: Inverse design for self-assembly via on-the-fly optimization. J. Chem. Phys. 2016, 145, 111101. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; van Anders, G.; Dodd, P.M.; Dshemuchadse, J.; Glotzer, S.C. Engineering entropy for the inverse design of colloidal crystals from hard shapes. Sci. Adv. 2019, 5, eaaw0514. [Google Scholar] [CrossRef] [Green Version]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocha, B.C.; Paul, S.; Vashisth, H. Role of Entropy in Colloidal Self-Assembly. Entropy 2020, 22, 877. https://doi.org/10.3390/e22080877
Rocha BC, Paul S, Vashisth H. Role of Entropy in Colloidal Self-Assembly. Entropy. 2020; 22(8):877. https://doi.org/10.3390/e22080877
Chicago/Turabian StyleRocha, Brunno C., Sanjib Paul, and Harish Vashisth. 2020. "Role of Entropy in Colloidal Self-Assembly" Entropy 22, no. 8: 877. https://doi.org/10.3390/e22080877
APA StyleRocha, B. C., Paul, S., & Vashisth, H. (2020). Role of Entropy in Colloidal Self-Assembly. Entropy, 22(8), 877. https://doi.org/10.3390/e22080877