
TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures

 , ,
, ,
Abstract
:1. Introduction

2. Data and Methods
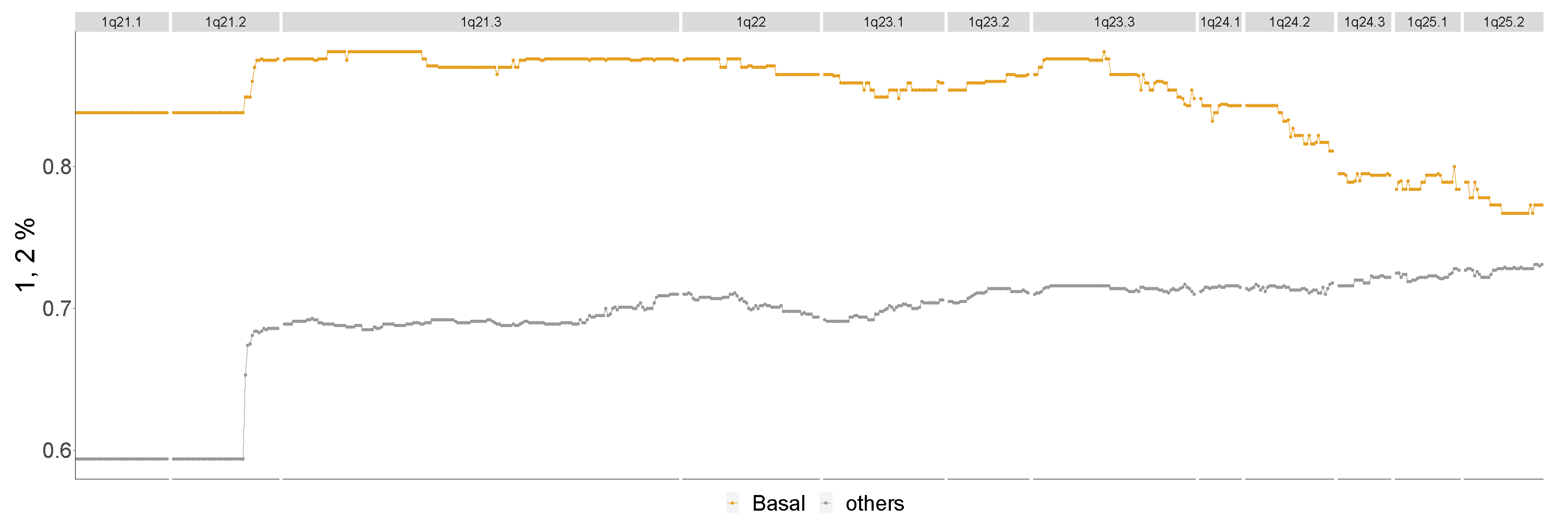
2.1. Persistence Curves
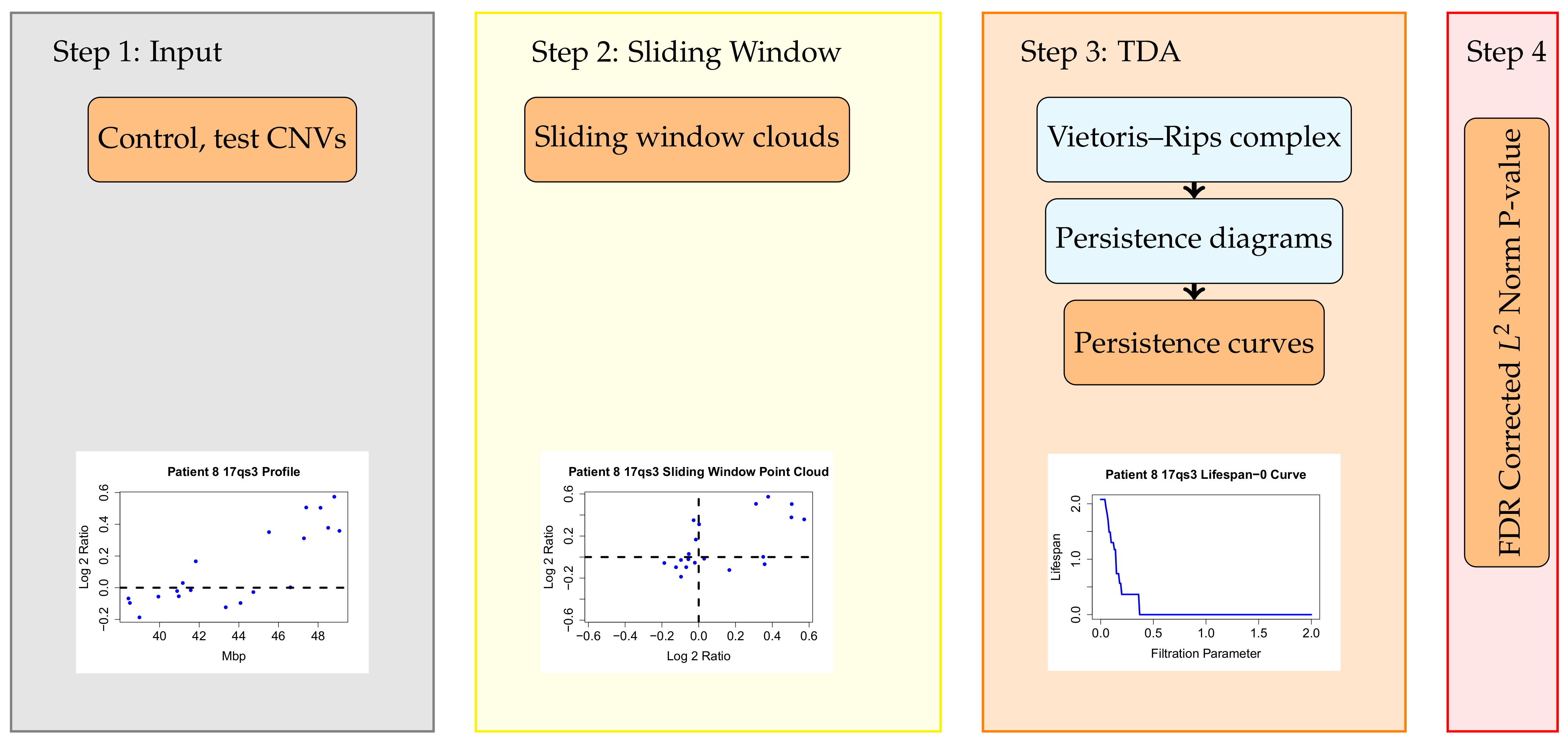
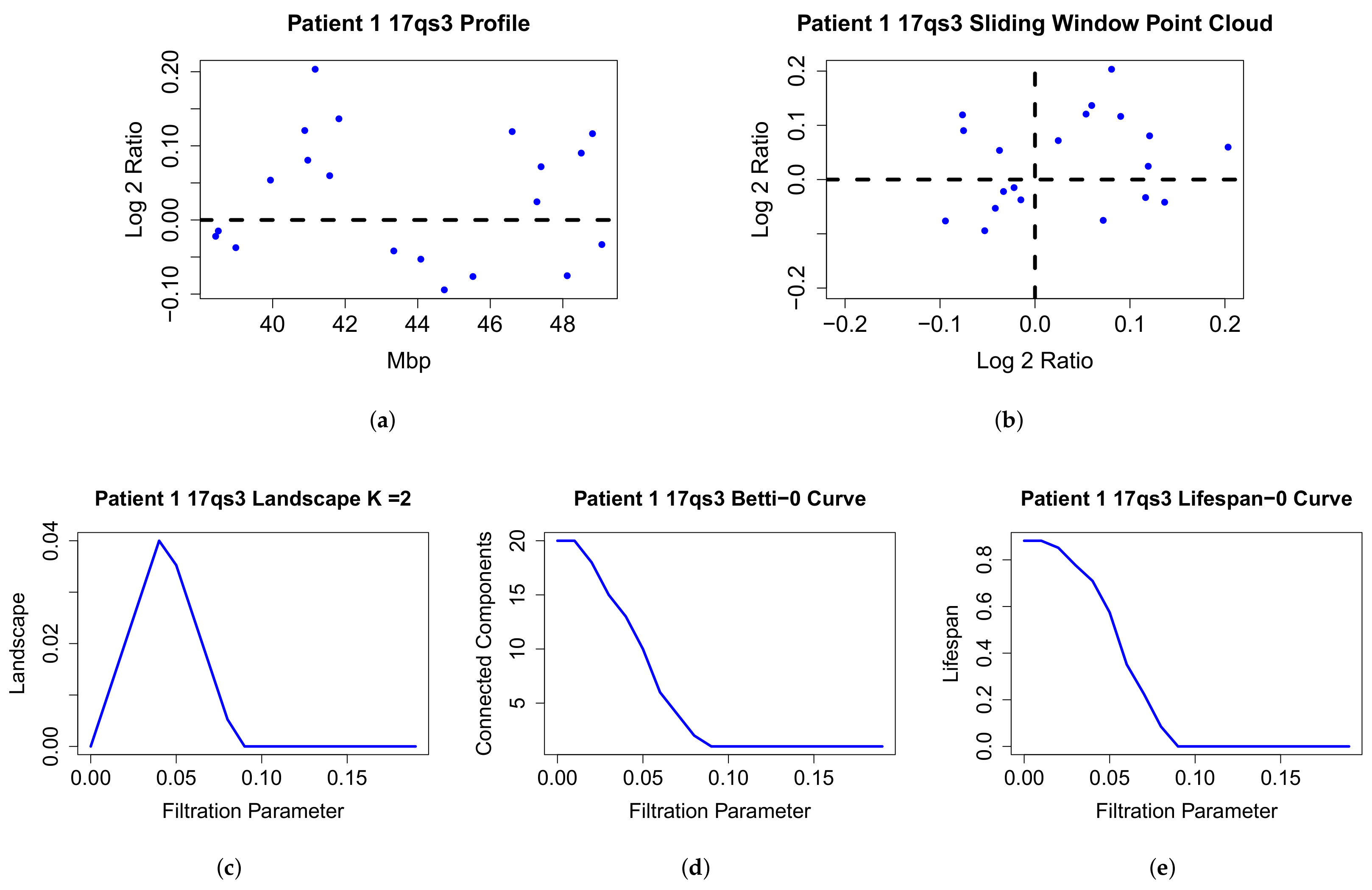
2.2. The Topological Analysis for Array CGH (TAaCGH) Method
2.3. Horlings Dataset
2.4. TCGA BRCA Cohort Data
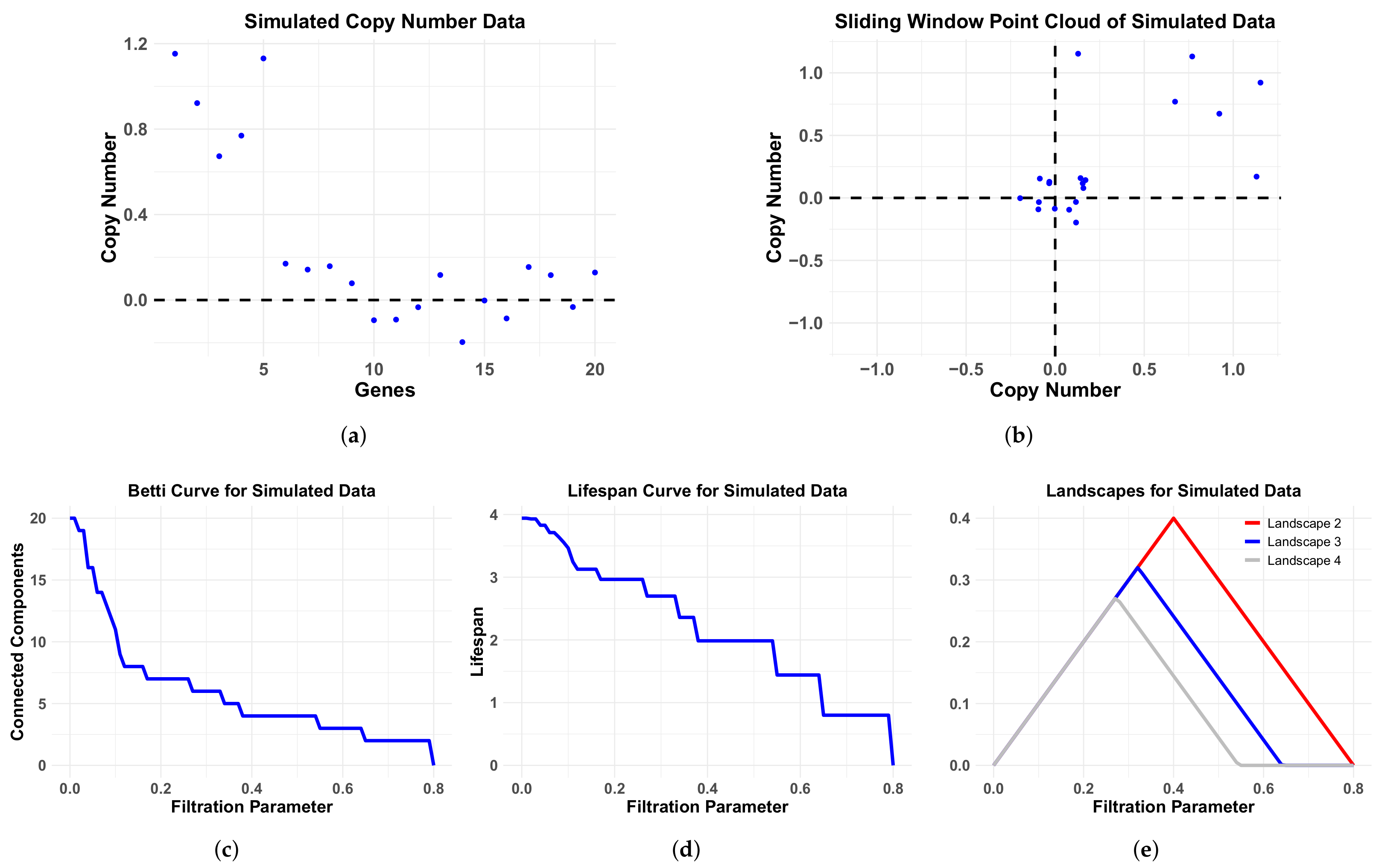
2.5. Simulation Data
2.6. Cancer Subtype Predictive Models
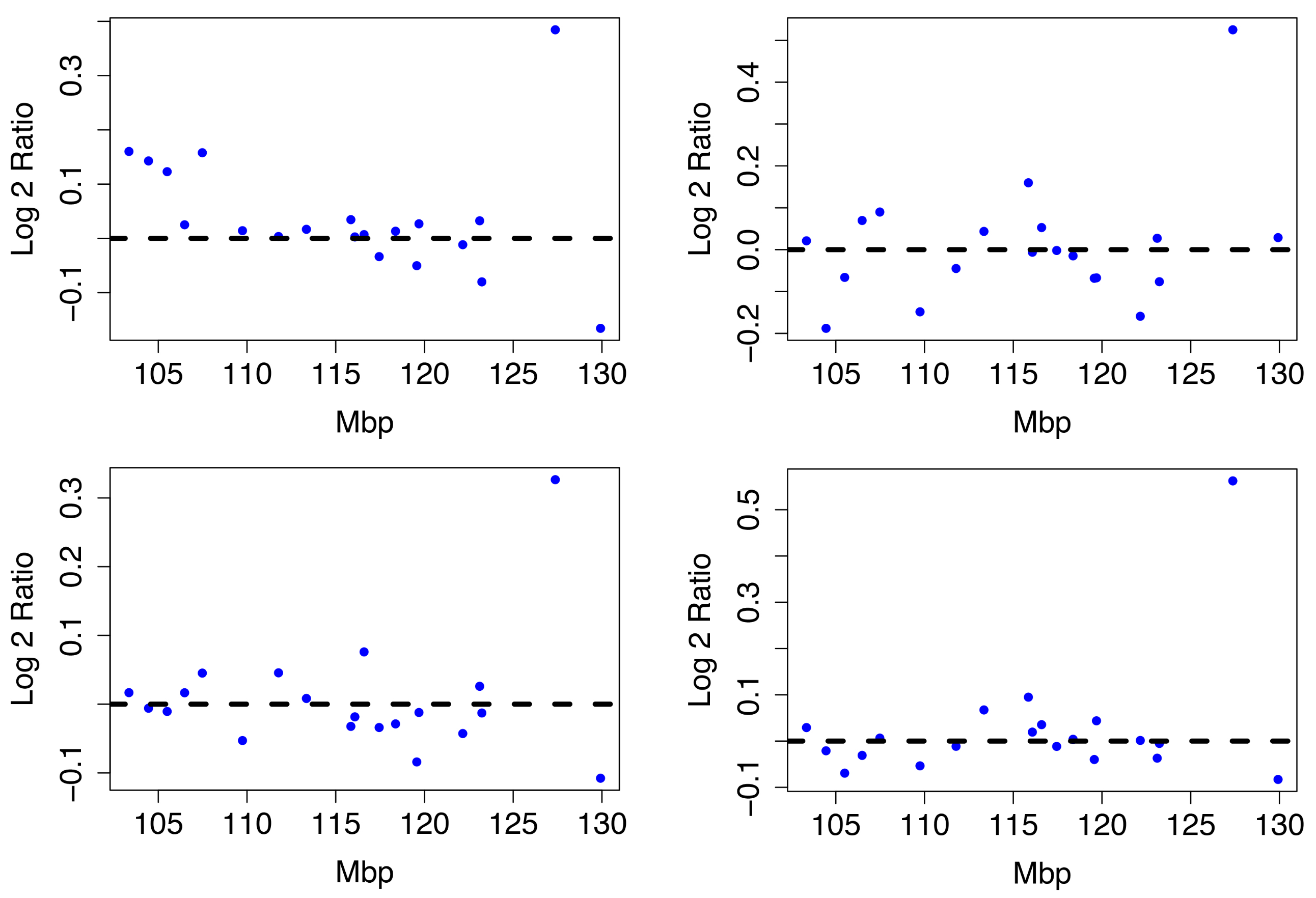
3. Bounds on the Distance between Persistence Curves
4. Computational Results
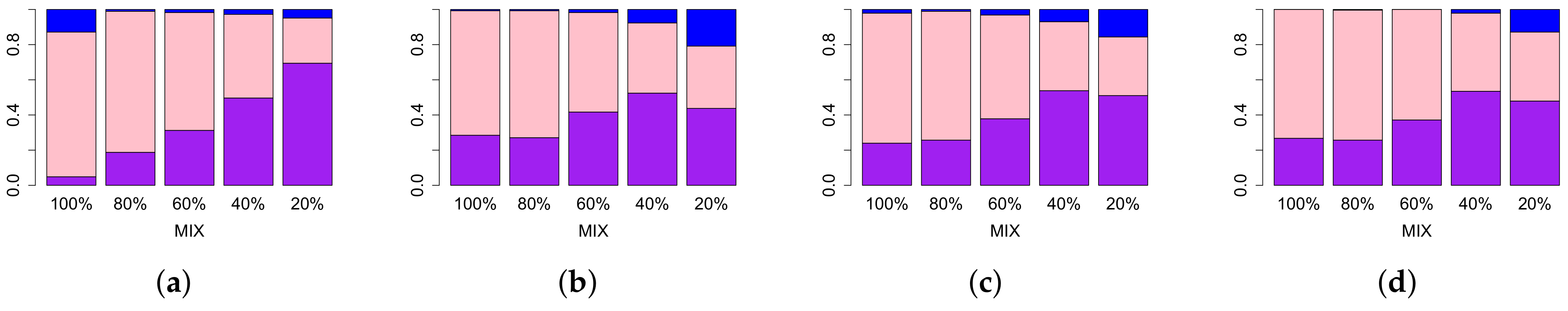
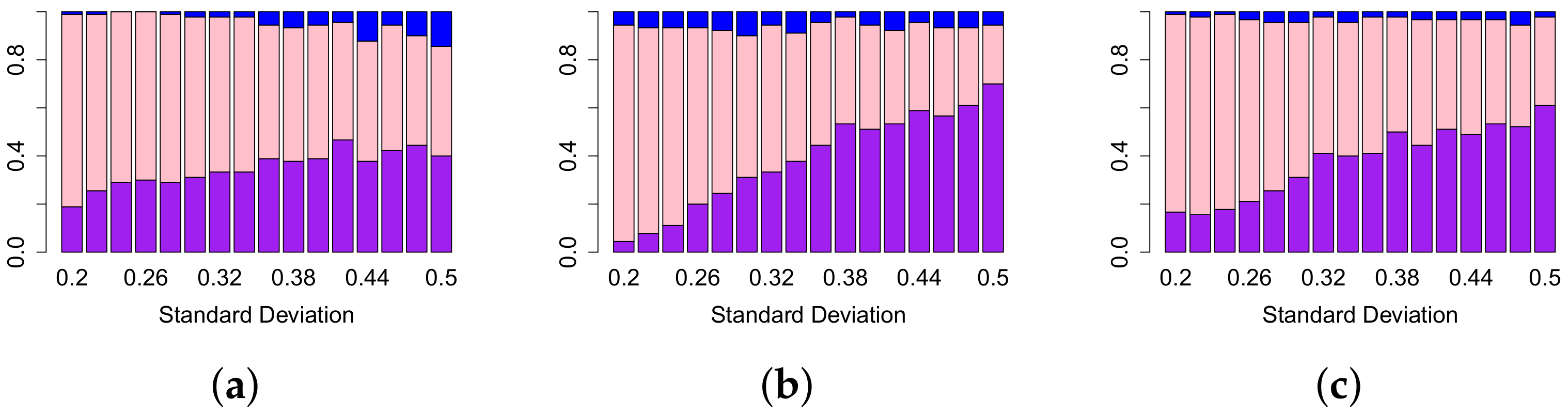
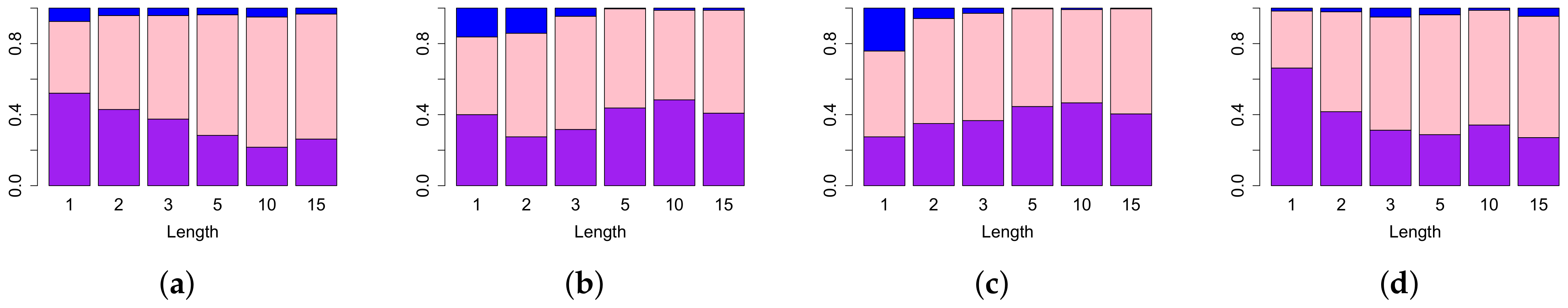
4.1. Comparison of Performance of Different Persistence Curves on Simulated Data
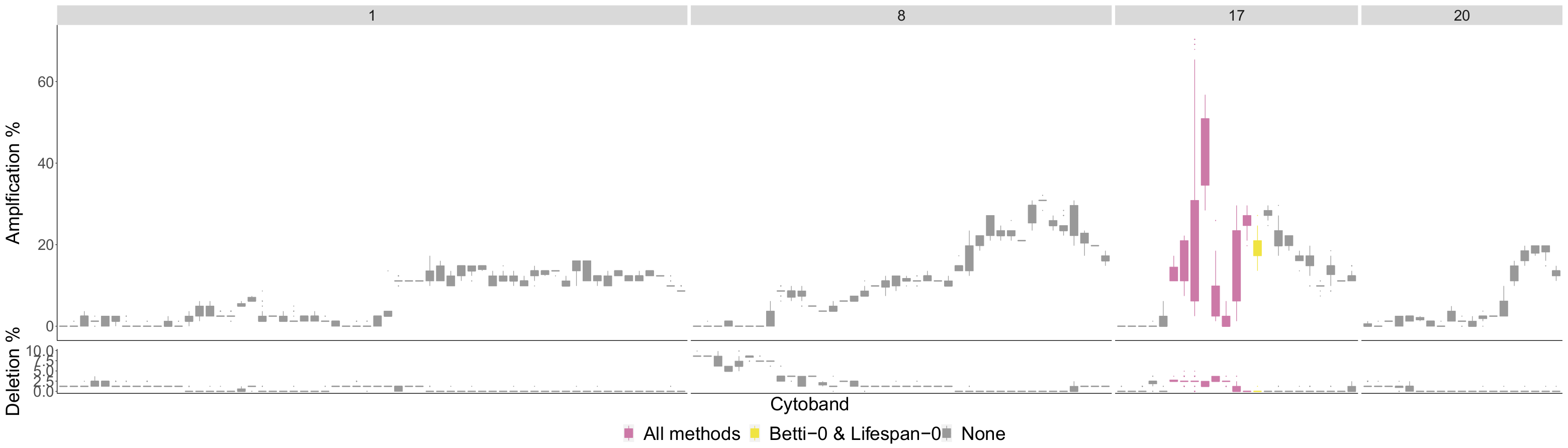
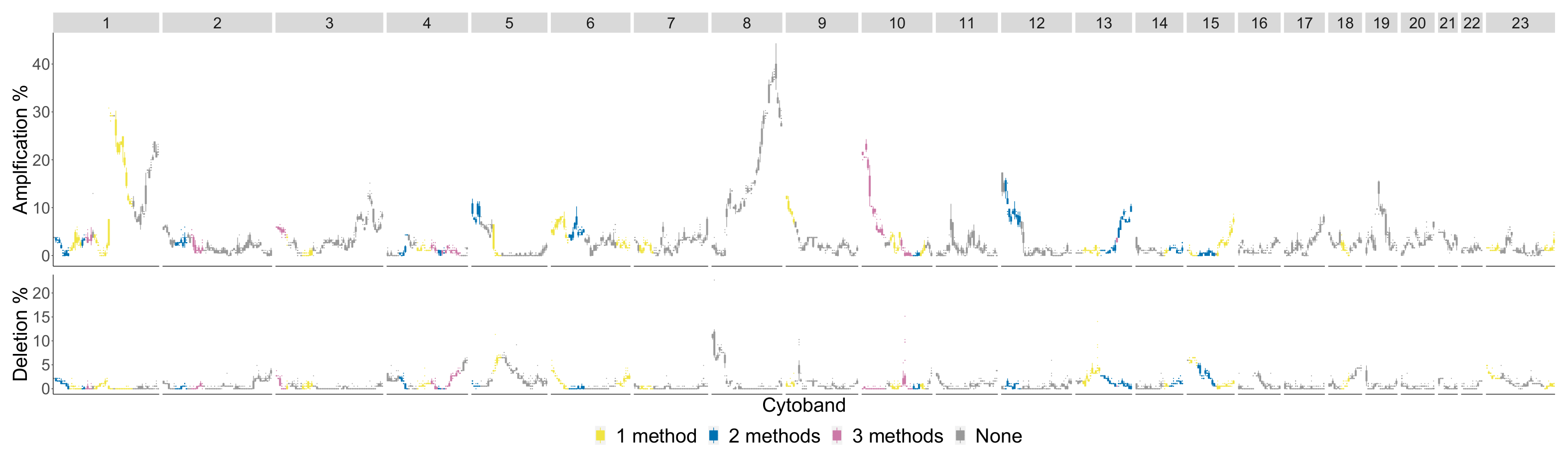
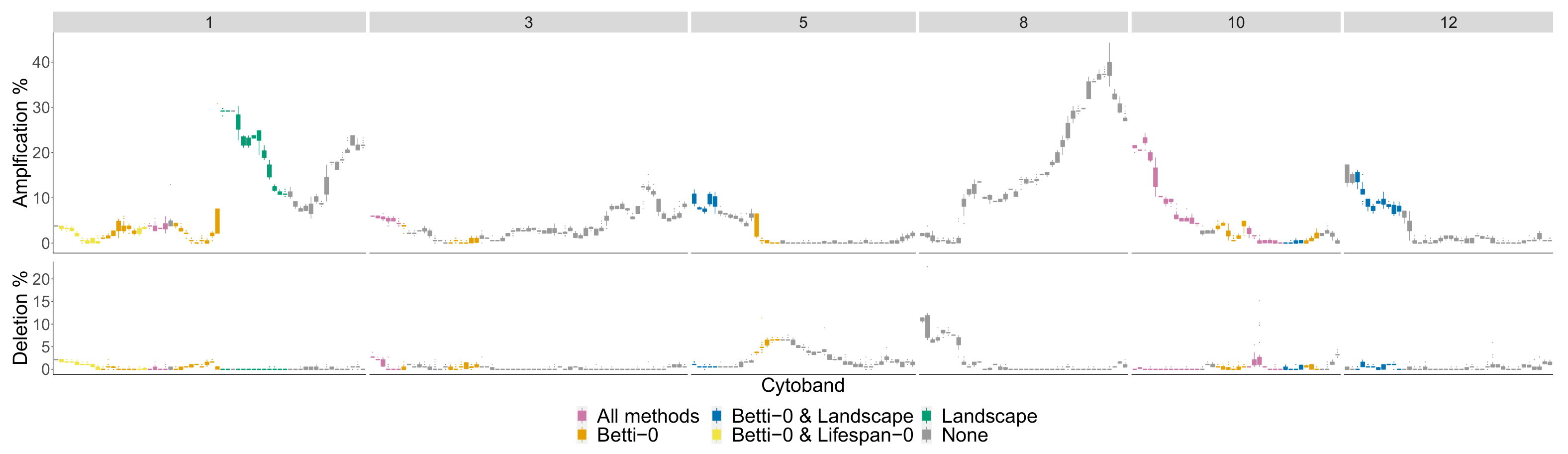
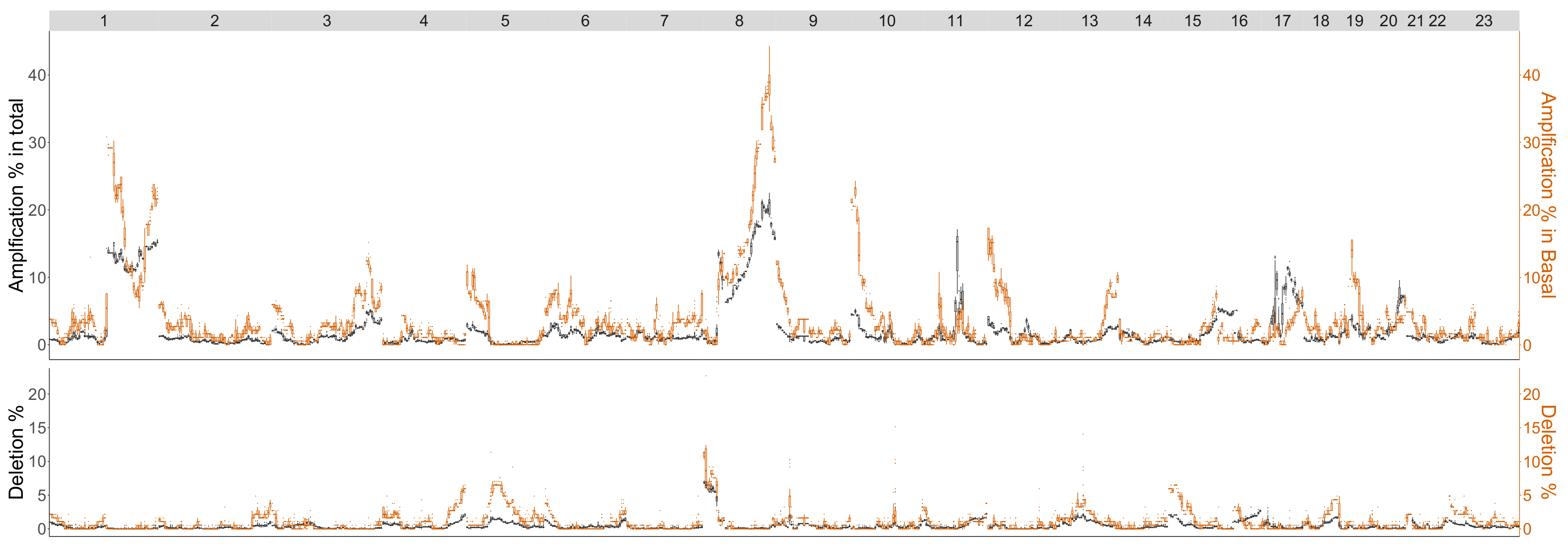
4.2. Comparison of Topological Summaries within the TAaCGH Framework on Horlings Data
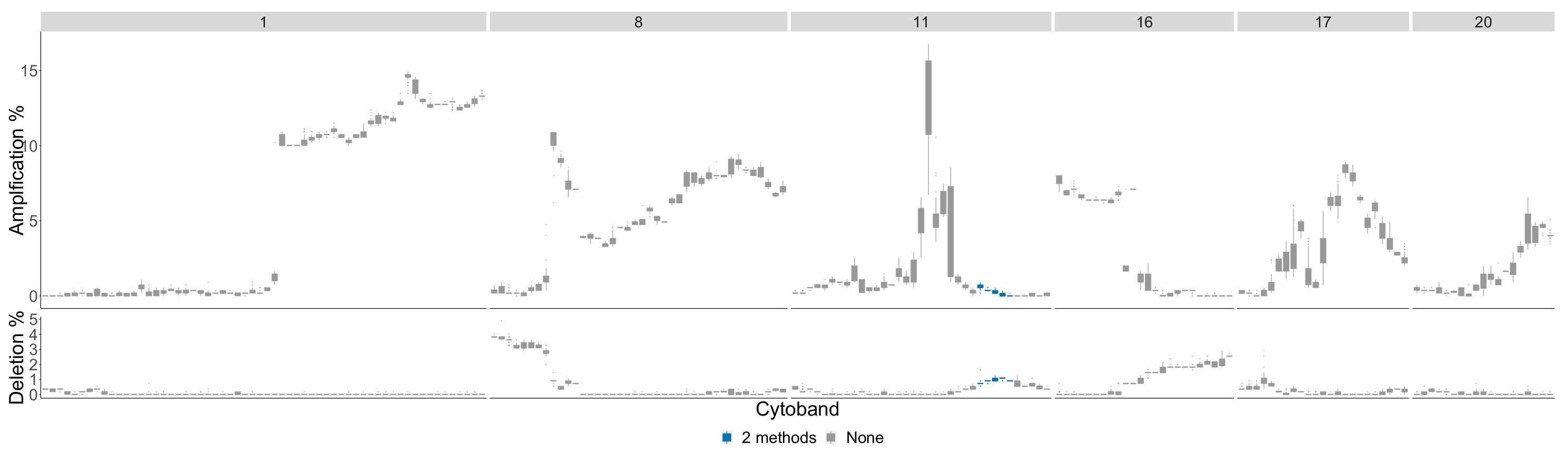
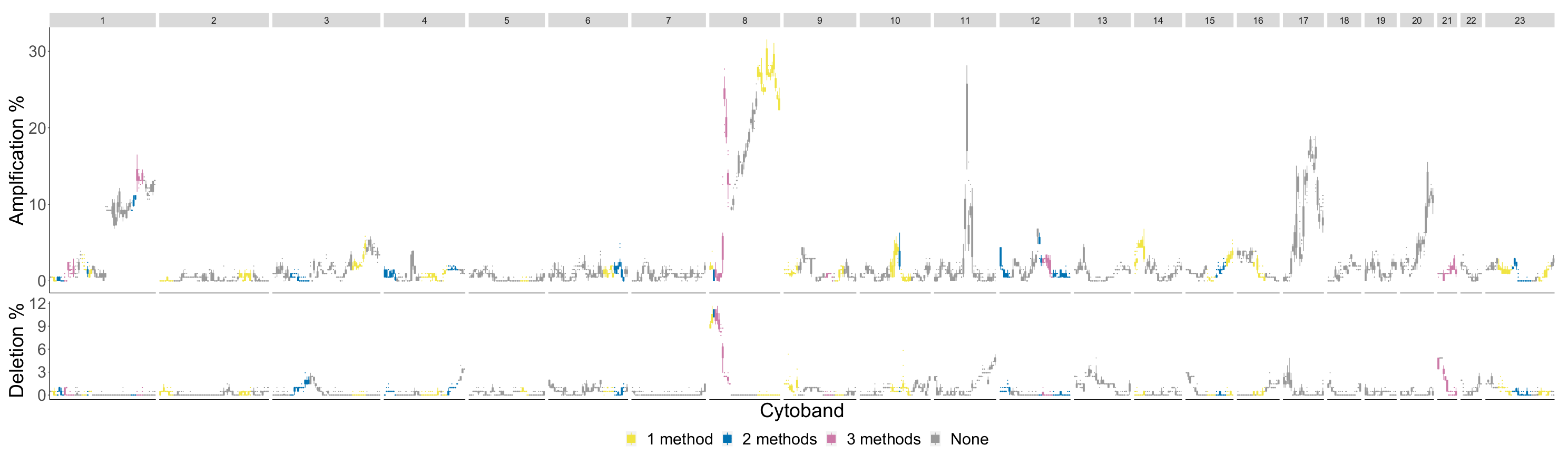
4.3. Detecting Breast Cancer Subtypes
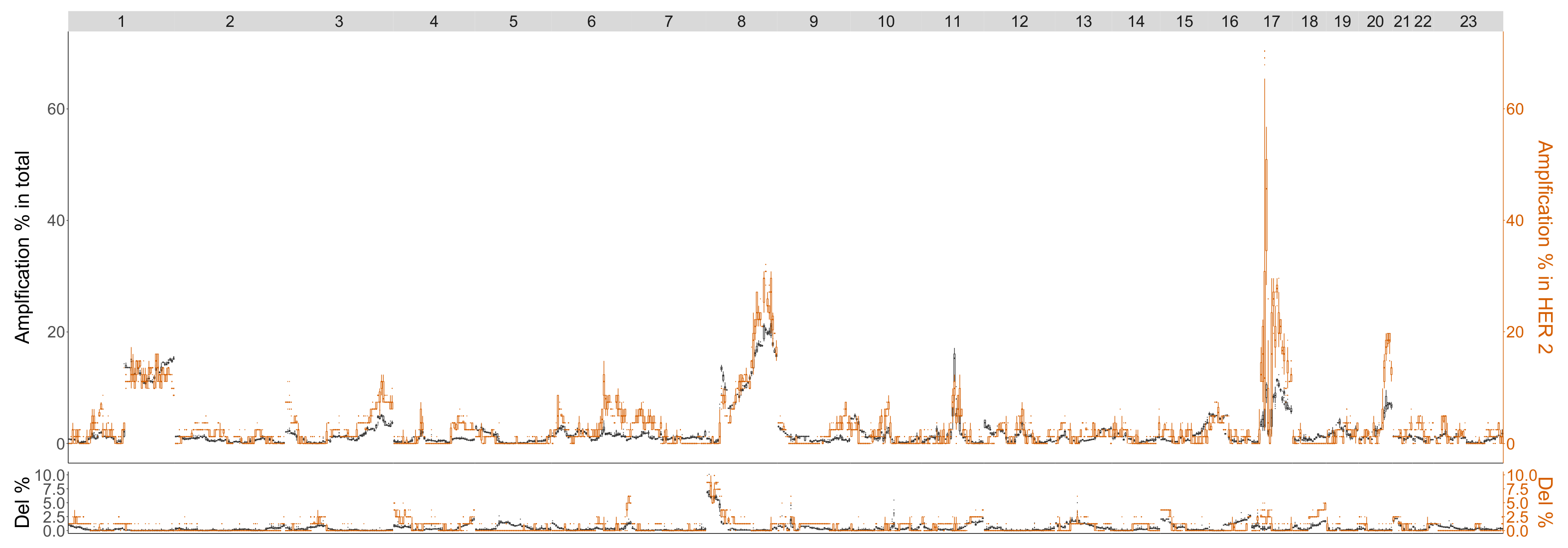
4.3.1. HER2
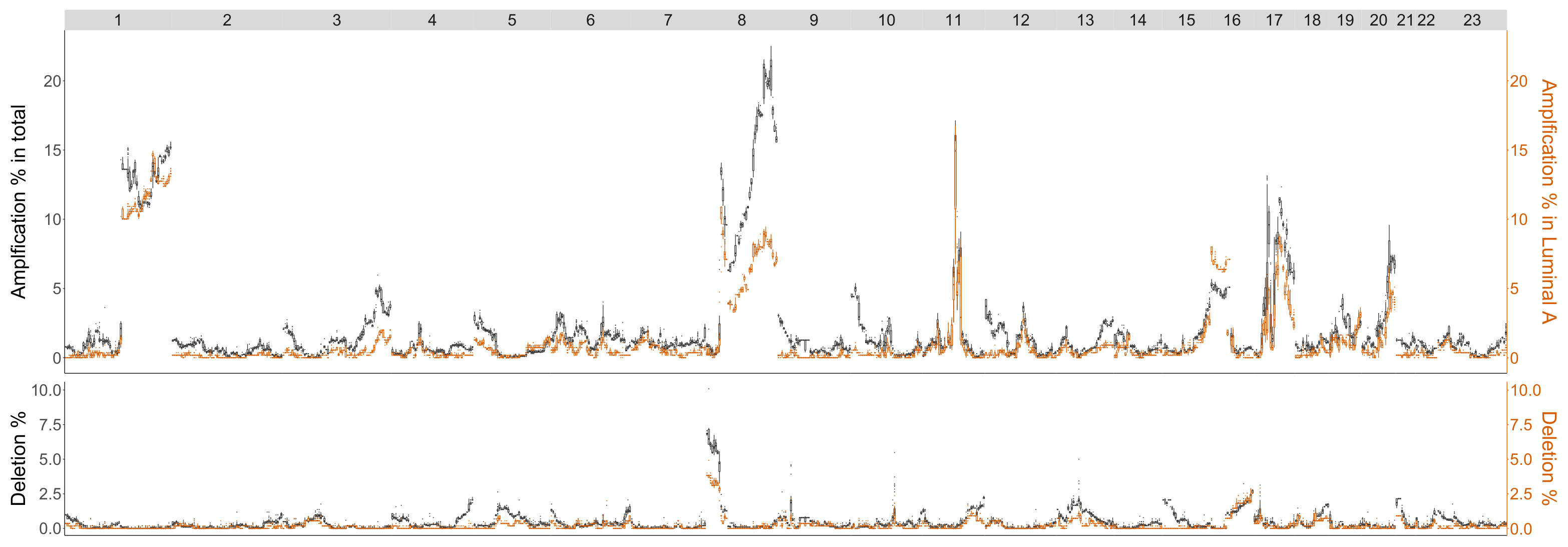
4.3.2. Luminal A
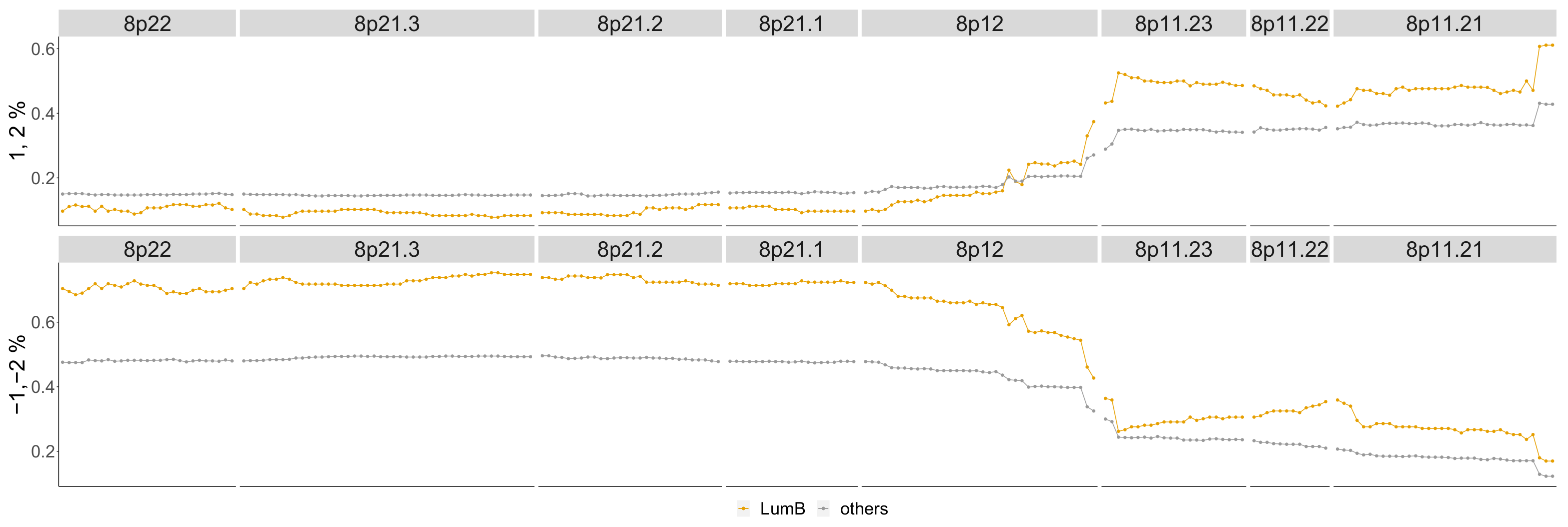
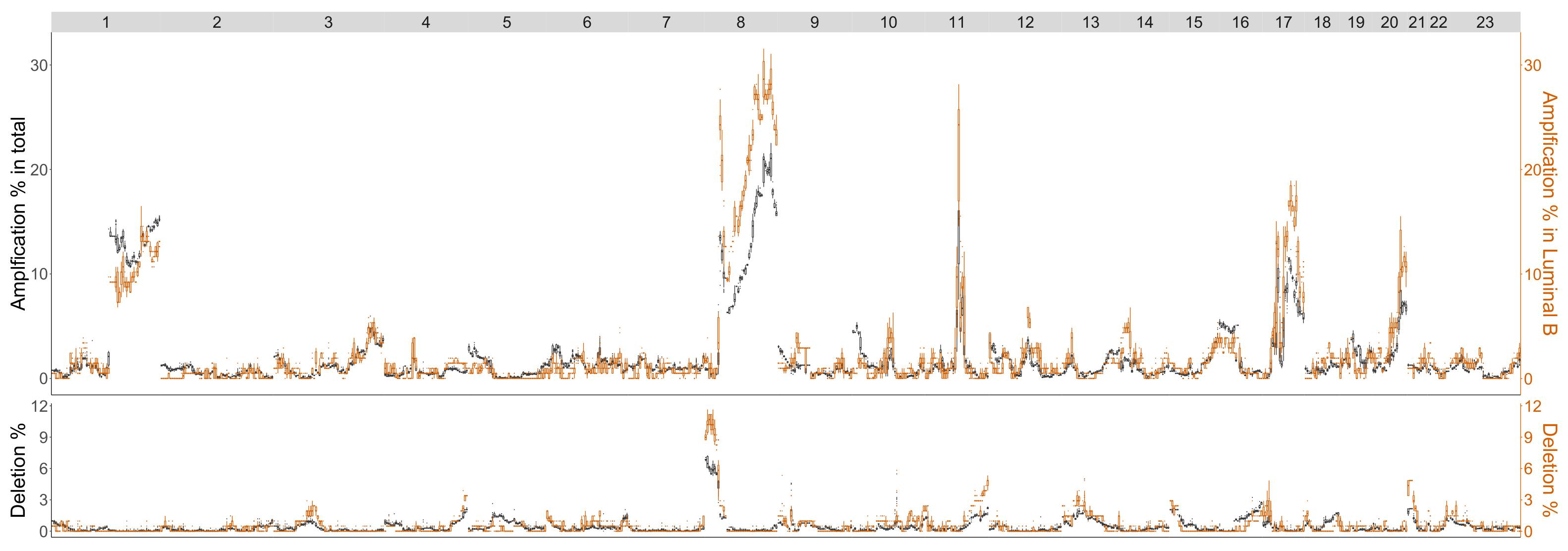
4.3.3. Luminal B
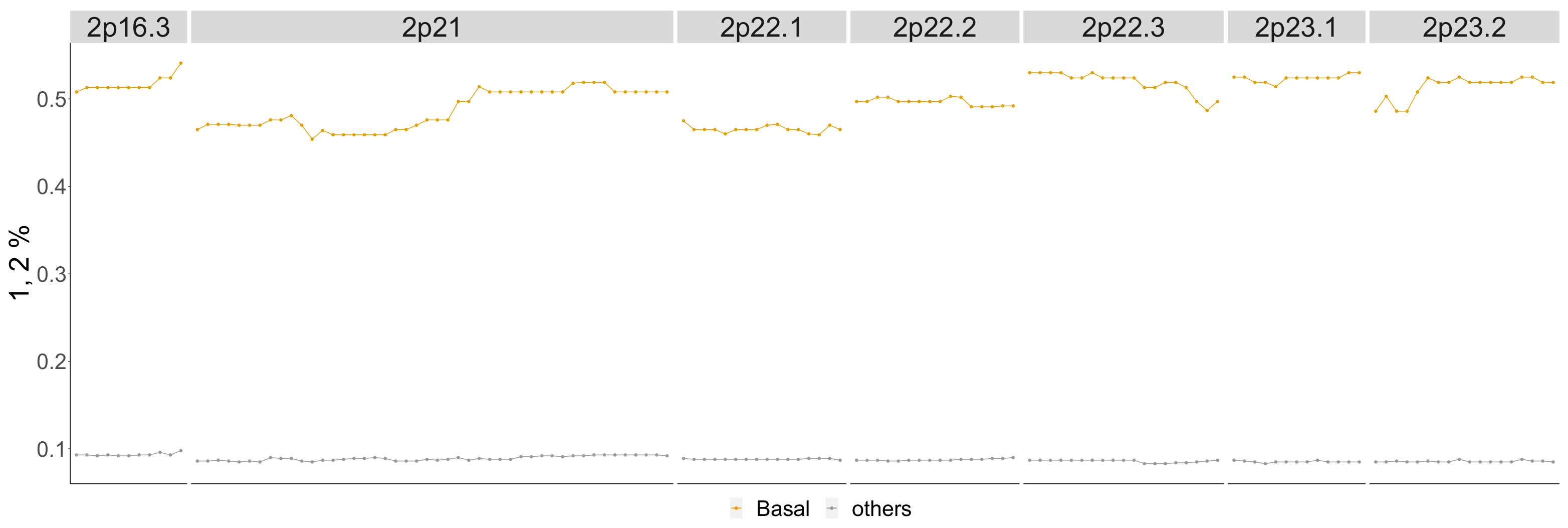
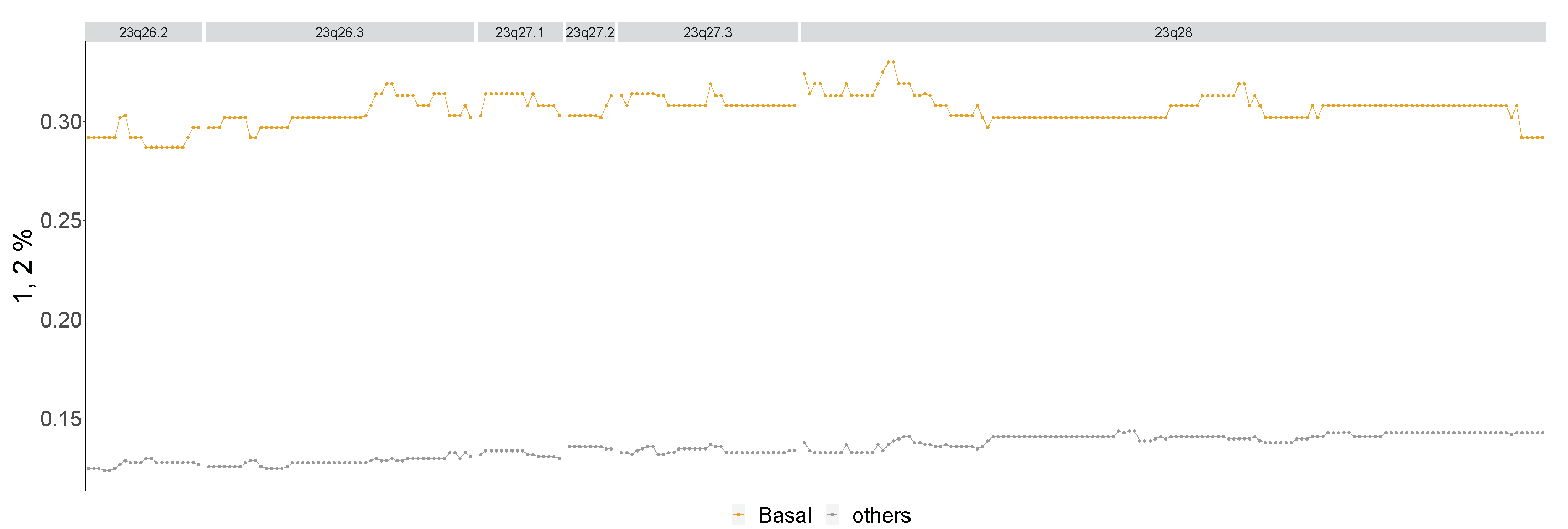
4.3.4. Basal
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Previously Detected Segments Not Detected with R TDA | |
|---|---|
| Basal | 1p34.2-p32.1, 1q23.1-q31.1, 3p25.1-p23, 3p22.1-p14.3, 3p14.2-p11.2, 4q13.3-q22.1, 4q32.3-q35.2, 5p15.2-p12, 9p23-p21.3, 9q32-q34.3, 10q11.21-q21.2, 10q25.2-q26.3, 12p13.33-p12.3, 14q12-q21.3, 18q11.1-q12.3, 18q12.3-q23 |
| Luminal B | 1p35.1-p33, 1q41-q44, 4q24-q27, 8p23.3-p22, 9p22.2-p21.1, 9q13-q22.1, |
| (No Basal in Control) | 13q12.2-q21.1, 13q31.1-q32.2 |
| HER2 | 17q21.2-q21.33 |
| Luminal B (No Basal in Control) | |
|---|---|
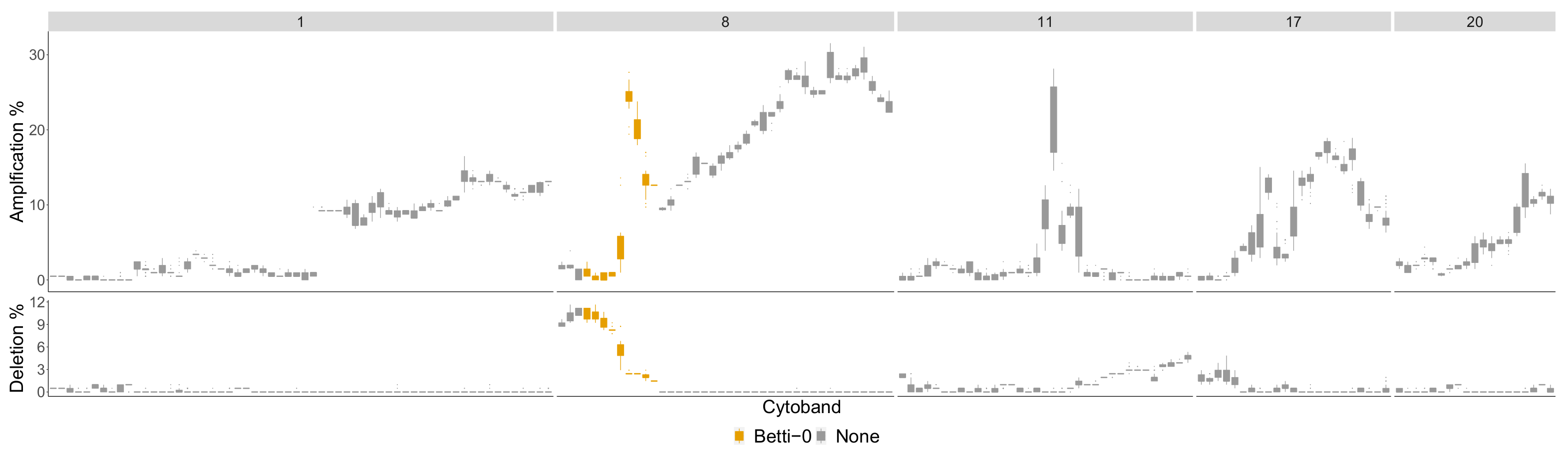
| Betti-0 | 1p36.32-p34.2, 1q32.1-q41, 8p22-p11.1, 8q24.11-q24.3, 9p24.3-p21.3, 9q21.33-q22.32, 9q31.1-q33.1, 12q21.31-q23.2, 21q11.2-q22.3, |
| Lifespan-0 | 1p36.11-p34.2, 1p31.1-p22.2, 1q31.1-q41, 1q32.1-q41, 3p22.3-p13, 3q24-q26.2, 4p16.3-p15.2, 4q31.3-q34.1, 6q24.1-q25.3, 8p23.3-p22, 8p22-p11.1, 9q21.33-q22.32, 10q23.1-q24.2, 12p13.33-p12.3, 12q21.1-q24.33, 15q23-q26.3, 21q11.2-q22.3, 23q11.1-q21.33 |
| Landscape | 1p32.1-p31.1, 2q31.1-q32.2, 3p22.1-p13, 4q31.21-q34.1, 5q23.1-q31.2, 6q22.31-q24.1, 12q21.31-q24.11, 21q11.2-q22.3, 23q11.1-q21.33 1p32.1-p31.1, 1q31.1-q41, 2p25.3-p23.2, 2q31.1-q33.1, 3p22.3-p13, 4p16.3-p15.2, 4q22.1-q25, 4q25-q28.3, 4q31.21-q34.1, 5q23.1-q31.2, 6q22.31-q24.1, 8p22-p11.1, 10q21.2-q23.1, 12q21.31-q24.11, 12q24.11-q24.33, 15q21.3-q25.2, 16q11.2-q21, 21q11.2-q22.3, 23p21.3-p11.21, 23q11.1-q12.3, 23q24-q27.2 1p36.22-p34.2, 1q31.1-q41, 3p22.1-p14.3, 4p16.3-p15.2, 6q24.1-q25.3, 8p23.1-p12, 8p22-p11.1, 8q22.1-q23.3, 9q21.33-q22.32, 10q21.2-q23.1, 12p13.33-p12.3, 12q21.1-q24.23, 14q11.2-q21.1, 16q12.2-q22.1 |











| Betti Curves | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 20% mix | 40% mix | 60% mix | 80% mix | 100% mix | ||||||
| 43.00% | 71.00% | 50.00% | 83.00% | 63.00% | 93.00% | 81.00% | 97.00% | 99.00% | 99.00% | |
| 52.00% | 60.00% | 58.00% | 67.00% | 66.00% | 73.00% | 73.00% | 77.00% | 82.00% | 83.00% | |
| Total | 47.50% | 65.50% | 54.00% | 75.00% | 64.50% | 83.00% | 77.00% | 87.00% | 90.50% | 91.00% |
| TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | |
| Landscape 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 20% mix | 40% mix | 60% mix | 80% mix | 100% mix | ||||||
| 26.00% | 93.00% | 41.00% | 97.00% | 59.00% | 99.00% | 76.00% | 99.00% | 90.00% | 100.00% | |
| 45.00% | 60.00% | 50.00% | 69.00% | 55.00% | 71.00% | 61.00% | 74.00% | 66.00% | 77.00% | |
| Total | 35.50% | 76.50% | 45.50% | 83.00% | 57.00% | 85.00% | 68.50% | 86.50% | 78.00% | 88.50% |
| TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | |
| Landscape 4 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 20% mix | 40% mix | 60% mix | 80% mix | 100% mix | ||||||
| 24.00% | 94.00% | 40.00% | 99.00% | 58.00% | 99.00% | 73.00% | 100.00% | 86.00% | 100.00% | |
| 45.00% | 64.00% | 51.00% | 71.00% | 58.00% | 75.00% | 66.00% | 78.00% | 72.00% | 81.00% | |
| Total | 34.50% | 79.00% | 45.50% | 85.00% | 58.00% | 87.00% | 69.50% | 89.00% | 79.00% | 90.50% |
| TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | |
References
- Ghoussaini, M.; Pharoah, P.D. Polygenic susceptibility to breast cancer: Current state-of-the-art. Future Oncol. 2009, 5, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Mars, N.; Widén, E.; Kerminen, S.; Meretoja, T.; Pirinen, M.; della Briotta Parolo, P.; Palta, P.; Palotie, A.; Kaprio, J.; Joensuu, H.; et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 2020, 11, 6383. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Coughlin, C.R.; Scharer, G.H.; Shaikh, T.H. Clinical impact of copy number variation analysis using high-resolution microarray technologies: Advantages, limitations and concerns. Genome Med. 2012, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, C.; Woodgett, J. Clinical uses of microarrays in cancer research. In Clinical Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2008; Volume 141, pp. 87–113. [Google Scholar]
- Cooper, C.S. Applications of microarray technology in breast cancer research. Breast Cancer Res. 2001, 3, 158. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Mol. Genet. 2009, 18, R163–R168. [Google Scholar] [CrossRef]
- DeWoskin, D.; Climent, J.; Cruz-White, I.; Vazquez, M.; Park, C.; Arsuaga, J. Applications of computational homology to the analysis of treatment response in breast cancer patients. Topol. Its Appl. 2010, 157, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Nicolau, M.; Levine, A.J.; Carlsson, G. Topology based data analysis identifies a subgroup of breast cancers with a unique mutational profile and excellent survival. Proc. Natl. Acad. Sci. USA 2011, 108, 7265–7270. [Google Scholar] [CrossRef] [Green Version]
- Rabadán, R.; Mohamedi, Y.; Rubin, U.; Chu, T.; Alghalith, A.N.; Elliott, O.; Arnés, L.; Cal, S.; Obaya, Á.J.; Levine, A.J.; et al. Identification of relevant genetic alterations in cancer using topological data analysis. Nat. Commun. 2020, 11, 3808. [Google Scholar]
- Climent, J.; Garcia, J.; Mao, J.; Arsuaga, J.; Perez-Losada, J. Characterization of breast cancer by array comparative genomic hybridization. Biochem. Cell Biol. 2007, 85, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Olshen, A.B.; Venkatraman, E.; Lucito, R.; Wigler, M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004, 5, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Arsuaga, J.; Borrman, T.; Cavalcante, R.; Gonzalez, G.; Park, C. Identification of copy number aberrations in breast cancer subtypes using persistence topology. Microarrays 2015, 4, 339–369. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Ushakova, A.; Sazdanovic, R.; Arsuaga, J. Prediction in Cancer Genomics Using Topological Signatures and Machine Learning. In Topological Data Analysis; Springer Nature: Cham, Switzerland, 2020; pp. 247–276. [Google Scholar]
- Ardanza-Trevijano, S.; Gonzalez, G.; Borrman, T.; Garcia, J.L.; Arsuaga, J. Topological analysis of amplicon structure in Comparative Genomic Hybridization (CGH) data: An application to ERBB2/HER2/NEU amplified tumors. In Computational Topology in Image Context. CTIC 2016; Lecture Notes in Computer Science; Bac, A., Mari, J.L., Eds.; Springer: Cham, Switzerland, 2016; Volume 9667. [Google Scholar] [CrossRef]
- Arriola, E.; Marchio, C.; Tan, D.S.; Drury, S.C.; Lambros, M.B.; Natrajan, R.; Rodriguez-Pinilla, S.M.; Mackay, A.; Tamber, N.; Fenwick, K.; et al. Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab. Investig. 2008, 88, 491–503. [Google Scholar] [CrossRef] [Green Version]
- Leiserson, M.D.; Vandin, F.; Wu, H.T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M.; et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Xian, L.; Adams, H.; Topaz, C.M.; Ziegelmeier, L. Capturing dynamics of time-varying data via topology. arXiv 2022, arXiv:2010.05780. [Google Scholar] [CrossRef]
- Chung, Y.M.; Lawson, A. Persistence curves: A canonical framework for summarizing persistence diagrams. Adv. Comput. Math. 2022, 48, 6. [Google Scholar] [CrossRef]
- Carrière, M.; Chazal, F.; Ike, Y.; Lacombe, T.; Royer, M.; Umeda, Y. Perslay: A neural network layer for persistence diagrams and new graph topological signatures. In Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics, Online. 26–28 August 2020; Volume 108, pp. 2786–2796. [Google Scholar]
- Horlings, H.M.; Lai, C.; Nuyten, D.S.; Halfwerk, H.; Kristel, P.; van Beers, E.; Joosse, S.A.; Klijn, C.; Nederlof, P.M.; Reinders, M.J.; et al. Integration of DNA copy number alterations and prognostic gene expression signatures in breast cancer patients. Clin. Cancer Res. 2010, 16, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Atienza, N.; Gonzalez-Diaz, R.; Soriano-Trigueros, M. A new entropy based summary function for topological data analysis. Electron. Notes Discret. Math. 2018, 68, 113–118. [Google Scholar] [CrossRef]
- Reininghaus, J.; Huber, S.; Bauer, U.; Kwitt, R. A stable multi-scale kernel for topological machine learning. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 7–12 June 2015; pp. 4741–4748. [Google Scholar]
- Bubenik, P. Statistical topological data analysis using persistence landscapes. J. Mach. Learn. Res. 2015, 16, 77–102. [Google Scholar]
- Adams, H.; Emerson, T.; Kirby, M.; Neville, R.; Peterson, C.; Shipman, P.; Chepushtanova, S.; Hanson, E.; Motta, F.; Ziegelmeier, L. Persistence images: A stable vector representation of persistent homology. J. Mach. Learn. Res. 2017, 18, 218–252. [Google Scholar]
- Perea, J.A.; Harer, J. Sliding windows and persistence: An application of topological methods to signal analysis. Found. Comput. Math. 2015, 15, 799–838. [Google Scholar] [CrossRef] [Green Version]
- Adams, H.; Tausz, A.; Vejdemo-Johansson, M. JavaPlex: A research software package for persistent (co) homology. In Proceedings of the International Congress on Mathematical Software, Lecture Notes in Computer Science, Seoul, Korea, 5–9 August 2014; Springer: Cham, Switzerland, 2014; Volume 8592, pp. 129–136. [Google Scholar]
- Broad GDAC Firehose. Available online: https://gdac.broadinstitute.org/ (accessed on 15 June 2022).
- Cohen-Steiner, D.; Edelsbrunner, H.; Harer, J. Stability of persistence diagrams. Discret. Comput. Geom. 2007, 37, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef] [Green Version]
- Moon, D.; Harer, J.; Bar-On, R. Maximum Number of Nonzero Persistence Cycles in a Vietoris–Rips Filtration. Private Communication. Available online: https://plv.colorado.edu/dmoon/assets/docs/nzp.pdf (accessed on 15 June 2022).
- Aigner, M.; Ziegler, G.M.; Hofmann, K.H.; Erdos, P. Proofs from the Book; Springer: Berlin/Heidelberg, Germany, 2010; Volume 274. [Google Scholar]
- Goff, M. Extremal Betti Numbers of Vietoris–Rips Complexes. Discret. Comput. Geom. 2011, 46, 132–155. [Google Scholar] [CrossRef] [Green Version]
- Ades, F.; Zardavas, D.; Bozovic-Spasojevic, I.; Pugliano, L.; Fumagalli, D.; De Azambuja, E.; Viale, G.; Sotiriou, C.; Piccart, M. Luminal B breast cancer: Molecular characterization, clinical management, and future perspectives. J. Clin. Oncol. 2014, 32, 2794–2803. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Sinha, R.; Hoadley, K.A.; Jacobsen, A.S.; Reva, B.; Perou, C.M.; Sander, C.; Schultz, N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res. Treat. 2013, 141, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Cornen, S.; Guille, A.; Adélaïde, J.; Addou-Klouche, L.; Finetti, P.; Saade, M.R.; Manai, M.; Carbuccia, N.; Bekhouche, I.; Letessier, A.; et al. Candidate luminal B breast cancer genes identified by genome, gene expression and DNA methylation profiling. PLoS ONE 2014, 9, e81843. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Adélaïde, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67, 11565–11575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Kuchenbaecker, K.B.; Michailidou, K.; Mendoza-Fandino, G.A.; Nord, S.; Lilyquist, J.; Olswold, C.; Hallberg, E.; Agata, S.; Ahsan, H.; et al. Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat. Commun. 2016, 7, 11375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendoza-Fandiño, G.; Lyra, P.C.M.; Nepomuceno, T.C.; Harro, C.M.; Woods, N.T.; Li, X.; Rangel, L.B.; Carvalho, M.A.; Couch, F.J.; Monteiro, A.N. Two distinct mechanisms underlie estrogen-receptor-negative breast cancer susceptibility at the 2p23. 2 locus. Eur. J. Hum. Genet. 2022, 30, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Ikink, G.J.; Boer, M.; Bakker, E.R.; Hilkens, J. IRS4 induces mammary tumorigenesis and confers resistance to HER2-targeted therapy through constitutive PI3K/AKT-pathway hyperactivation. Nat. Commun. 2016, 7, 13567. [Google Scholar] [CrossRef] [Green Version]
- Ouban, A. Filamin-A expression in triple-negative breast cancer and its clinical significance. Biotechnol. Biotechnol. Equip. 2021, 35, 1409–1419. [Google Scholar] [CrossRef]
- Sabatier, R.; Finetti, P.; Adelaide, J.; Guille, A.; Borg, J.P.; Chaffanet, M.; Lane, L.; Birnbaum, D.; Bertucci, F. Down-regulation of ECRG4, a candidate tumor suppressor gene, in human breast cancer. PLoS ONE 2011, 6, e27656. [Google Scholar] [CrossRef] [Green Version]
- Dratwa, M.; Wysoczanska, B.; Brankiewicz, W.; Stachowicz-Suhs, M.; Wietrzyk, J.; Matkowski, R.; Ekiert, M.; Szelachowska, J.; Maciejczyk, A.; Szajewski, M.; et al. Relationship between Telomere Length, TERT Genetic Variability and TERT, TP53, SP1, MYC Gene Co-Expression in the Clinicopathological Profile of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 5164. [Google Scholar] [CrossRef]
- Da Silva, E.M.; Selenica, P.; Vahdatinia, M.; Pareja, F.; Da Cruz Paula, A.; Ferrando, L.; Gazzo, A.M.; Dopeso, H.; Ross, D.S.; Bakhteri, A.; et al. TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. NPJ Breast Cancer 2021, 7, 43. [Google Scholar] [CrossRef]
- Gay-Bellile, M.; Veronese, L.; Combes, P.; Eymard-Pierre, E.; Kwiatkowski, F.; Dauplat, M.M.; Cayre, A.; Privat, M.; Abrial, C.; Bignon, Y.J.; et al. TERT promoter status and gene copy number gains: Effect on TERT expression and association with prognosis in breast cancer. Oncotarget 2017, 8, 77540. [Google Scholar] [CrossRef]
- Panani, A.D. Isochromosome 5p, a novel recurrent abnormality in breast cancer: Is it a common abnormality in cancer? In Vivo 2010, 24, 715–717. [Google Scholar]
- Bartels, S.; van Luttikhuizen, J.L.; Christgen, M.; Mägel, L.; Luft, A.; Hänzelmann, S.; Lehmann, U.; Schlegelberger, B.; Leo, F.; Steinemann, D.; et al. CDKN2A loss and PIK3CA mutation in myoepithelial-like metaplastic breast cancer. J. Pathol. 2018, 245, 373–383. [Google Scholar] [CrossRef]













| Lifespan Curves | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 20% mix | 40% mix | 60% mix | 80% mix | 100% mix | ||||||
| 32.00% | 85.00% | 42.00% | 96.00% | 60.00% | 99.00% | 80.00% | 100.00% | 99.00% | 100.00% | |
| 49.00% | 62.00% | 56.00% | 70.00% | 63.00% | 76.00% | 71.00% | 79.00% | 78.00% | 84.00% | |
| Total | 40.50% | 73.50% | 49.00% | 83.00% | 61.50% | 87.50% | 75.50% | 89.50% | 88.50% | 92.00% |
| TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | |
| Landscape 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 20% mix | 40% mix | 60% mix | 80% mix | 100% mix | ||||||
| 27.00% | 92.00% | 41.00% | 98.00% | 59.00% | 99.00% | 78.00% | 100.00% | 93.00% | 100.00% | |
| 49.00% | 60.00% | 48.00% | 68.00% | 53.00% | 70.00% | 58.00% | 73.00% | 63.00% | 76.00% | |
| Total | 38.00% | 76.00% | 44.50% | 83.00% | 56.00% | 84.50% | 68.00% | 86.50% | 78.00% | 88.00% |
| TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | TPR | SPC | |
| HER | |
|---|---|
| Betti-0 | 17q 11.1-q21.31, 17q21.31-q22 |
| Lifespan-0 | 17q11.1-q22 |
| Landscape | : 17q11.1-q21.31, : 17q11.1-q21.33, : 17q11.1-q21.33 |
| Betti HER2 | Lifespan HER2 | Landscape 2 HER2 | Landscape 3 HER2 | Landscape 4 HER2 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 9 | 5 | 9 | 5 | 8 | 6 | 7 | 7 | 6 | 8 |
| 2 | 48 | 0 | 50 | 0 | 50 | 2 | 48 | 0 | 50 |
| Accuracy: 89% | Accuracy: 92% | Accuracy: 90% | Accuracy: 86% | Accuracy: 88% | |||||
| Luminal A | |
|---|---|
| Betti-0 | 11q 22.1-q23.2 |
| Lifespan-0 | 2q12.1-q21.1, 5p14.3-p12 |
| Landscape | : 2q12.1-q21.1, 5p14.3-p12, 11q22.1-q23.2 |
| Luminal A Lifespan | Luminal A Landscape 3 | ||
|---|---|---|---|
| 11 | 7 | 9 | 9 |
| 6 | 40 | 2 | 44 |
| Accuracy: 80% | Accuracy: 83% | ||
| Basal | |
|---|---|
| Betti-0 | 1p 36.32-p33, 1p32.3-p31.1, 1p22.2-p12, 2p23.2-p16.3, 2p15-p11.2, 3p26.3-p24.3, 3p21.2-p13, 4p15.1-p11, 4q21.21-q34.1 5p15.33-p15.1, 5q11.1-q13.1, 6p25.3-p22.1, 6p21.33-p11.2, 6q24.1-q27, 7p21.3-p14.2, 9p24.3-p22.3, 10p15.3-p11.1, 10q21.1-q22.1, 10q22.2-q26.11, 12p13.31-p11.21, 13q12.2-q31.2, 13q31.2-q34, 14q24.3-q32.33, 15q11.2-q22.31, 15q23-q26.3, 18q12.1-q21.2, 23p22.33-p11.21 |
| Lifespan-0 | 1p36.32-p36.11, 1p32.3-p31.1, 2p23.2-p16.3, 2p15-p11.2, 3p26.3-p25.1, 4q24-q27, 4q28.3-q31.3, 4q31.3-q34.1, 6p21.33-p11.2, 10p15.3-p11.1, 10q23.1-q24.2, 13q21.1-q31.2, 15q14-q22.31, 23p13.2-p12 |
| Landscape | 1p32.1-p31.1, 1q21.1-q25.2, 2p15-p11.2, 3p26.3-p25.1, 4p15.1-p11, 4q24-q28.3, 4q31.21-q34.1, 5p15.33-p15.1, 10p15.3-p12.31, 10p12.31-p11.1, 10q23.1-q25.1, 12p13.31-p11.21, 13q31.2-q34, 14q31.3-q32.33, 23p22.33-p21.3, 23q26.2-q28 |
| Betti Basal | Lifespan Basal | Basal | |||
|---|---|---|---|---|---|
| 17 | 2 | 14 | 5 | 16 | 3 |
| 3 | 42 | 4 | 1 | 6 | 39 |
| Accuracy: 92% | Accuracy: 86% | Accuracy: 86% | |||
| Cytoband Ranges of Newly Detected Segments | |
|---|---|
| Betti-0 | 2p23.2-p16.3, 8p22-p11.1 |
| Lifespan-0 | 2p23.2-p16.3, 2q12.1-q21.1, 5p14.3-p12 |
| Landscape | : 2q12.1-q21.1, 5p14.3-p12, : 1q21.1-q25.2, 23q26.2-q28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, J.; Ardanza-Trevijano, S.; Xiong, J.; Arsuaga, J.; Sazdanovic, R. TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures. Entropy 2022, 24, 896. https://doi.org/10.3390/e24070896
Aslam J, Ardanza-Trevijano S, Xiong J, Arsuaga J, Sazdanovic R. TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures. Entropy. 2022; 24(7):896. https://doi.org/10.3390/e24070896
Chicago/Turabian StyleAslam, Jai, Sergio Ardanza-Trevijano, Jingwei Xiong, Javier Arsuaga, and Radmila Sazdanovic. 2022. "TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures" Entropy 24, no. 7: 896. https://doi.org/10.3390/e24070896
APA StyleAslam, J., Ardanza-Trevijano, S., Xiong, J., Arsuaga, J., & Sazdanovic, R. (2022). TAaCGH Suite for Detecting Cancer—Specific Copy Number Changes Using Topological Signatures. Entropy, 24(7), 896. https://doi.org/10.3390/e24070896