

The Weight-Based Feature Selection (WBFS) Algorithm Classifies Lung Cancer Subtypes Using Proteomic Data


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. Proteome Profiling Analysis
2.3. Using WBFS to Obtain Candidate Protein Biomarkers
| Algorithm 1 Weight-based feature selection (WBFS) |
| ; |
2.4. Using Bayesian Networks to Discover Causalities
2.5. Receiver Operating Characteristic (ROC) and Survival Analysis
3. Results
3.1. The Distributions Overview of the LUAD and LUSC Tumor Samples
3.2. Using WBFS Method Identifying Protein Signatures for Classifying the Two Cancer Subtypes
3.2.1. Evaluate WBFS Classification Performance Based on UCI Datasets
3.2.2. Using WBFS to Obtain the Top 10 Candidate Biomarkers
3.3. Using Bayesian Networks to Discover Causalities
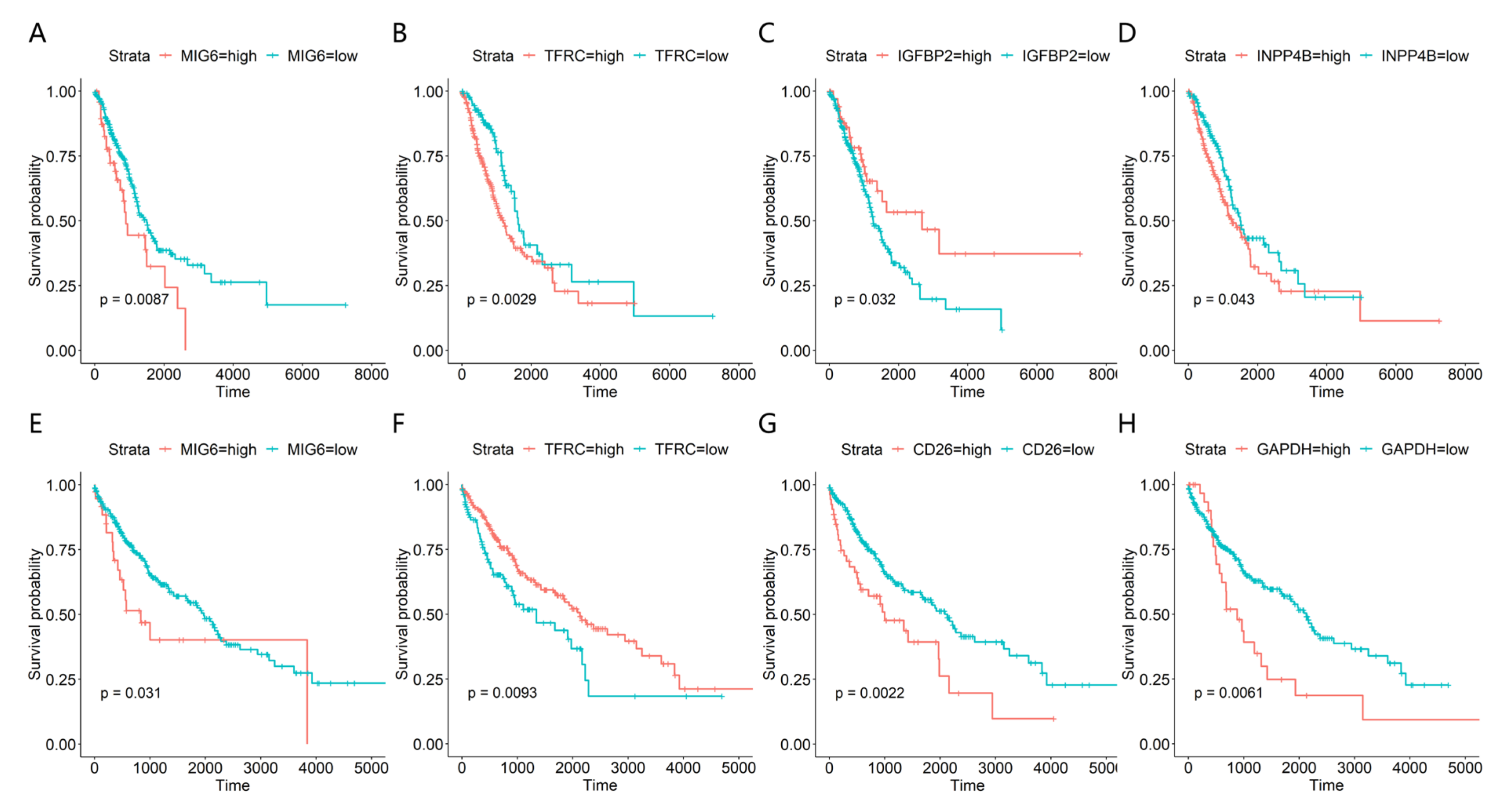
3.4. ROC and Survival Analysis of the Candidate Protein Signatures
4. Discussion
- (1)
- Developing a more efficient feature selection algorithm that can automatically determine the optimal number of selected features based on the intrinsic dimension of a high-dimensional dataset.
- (2)
- Investigating the relationship between causal and non-causal feature selection methods and applying non-causal feature selection methods to the Bayesian network. This will facilitate Bayesian network structure learning based on high-dimensional data.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Relli, V.; Trerotola, M.; Guerra, E.; Alberti, S. Abandoning the notion of non-small cell lung cancer. Trends Mol. Med. 2019, 25, 585–594. [Google Scholar] [CrossRef]
- Li, J.; Lu, Y.; Akbani, R.; Ju, Z.; Roebuck, P.L.; Liu, W.; Yang, J.-Y.; Broom, B.M.; Verhaak, R.G.W.; Kane, D.W.; et al. TCPA: A resource for cancer functional proteomics data. Nat. Methods 2013, 10, 1046–1047. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Zhu, Y.; Ji, A.; Zhang, Q.; Liao, G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci. Rep. 2020, 40, BSR20194337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Qu, J.; Cao, W.; Liu, Y.; Zheng, G.; Zhang, E.; Cai, Z. Identification of prognostic genes in the acute myeloid leukemia immune microenvironment based on TCGA data analysis. Cancer Immunol. Immunother. 2019, 68, 1971–1978. [Google Scholar] [CrossRef]
- Song, X.-F.; Zhang, Y.; Guo, Y.-N.; Sun, X.-Y.; Wang, Y.-L. Variable-size cooperative coevolutionary particle swarm optimization for feature selection on high-dimensional data. IEEE Trans. Evol. Comput. 2020, 24, 882–895. [Google Scholar] [CrossRef]
- Kumar, S.; Patnaik, S.; Dixit, A. Predictive models for stage and risk classification in head and neck squamous cell carcinoma (HNSCC). PeerJ 2020, 8, e9656. [Google Scholar] [CrossRef]
- Torres, R.; Judson-Torres, R.L. Research techniques made simple: Feature selection for biomarker discovery. J. Investig. Dermatol. 2019, 139, 2068–2074.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, J.R.; Estévez, P.A. A review of feature selection methods based on mutual information. Neural Comput. Appl. 2014, 24, 175–186. [Google Scholar] [CrossRef]
- Battiti, R. Using mutual information for selecting features in supervised neural net learning. IEEE Trans. Neural Netw. 1994, 5, 537–550. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.D. Feature Selection and Feature Extraction for Text Categorization. In Proceedings of the Speech and Natural Language: Proceedings of a Workshop Held at Harriman, Harriman, NY, USA, 23–26 February 1992. [Google Scholar]
- Kwak, N.; Choi, C.-H. Input feature selection for classification problems. IEEE Trans. Neural Netw. 2002, 13, 143–159. [Google Scholar] [CrossRef]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Tang, X. Conditional Infomax Learning: An Integrated Framework for Feature Extraction and Fusion. In European Conference on Computer Vision; Springer: Berlin/Heidelberg, Germany, 2006; pp. 68–82. [Google Scholar]
- Brown, G.; Pocock, A.; Zhao, M.-J.; Luján, M. Conditional likelihood maximisation: A unifying framework for information theoretic feature selection. J. Mach. Learn. Res. 2012, 13, 27–66. [Google Scholar]
- Wan, J.; Chen, H.; Li, T.; Yang, X.; Sang, B. Dynamic interaction feature selection based on fuzzy rough set. Inf. Sci. 2021, 581, 891–911. [Google Scholar] [CrossRef]
- Nakariyakul, S. A hybrid gene selection algorithm based on interaction information for microarray-based cancer classification. PLoS ONE 2019, 14, e0212333. [Google Scholar] [CrossRef]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Krijthe, J.; van der Maaten, L.; Krijthe, M.J.; Package ‘Rtsne’. R Package Version 0.13 2017URL. 2018. Available online: https://github.com/jkrijthe/Rtsne (accessed on 11 January 2023).
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Glymour, C.; Zhang, K.; Spirtes, P. Review of causal discovery methods based on graphical models. Front. Genet. 2019, 10, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Pollino, C.A. Good practice in Bayesian network modelling. Environ. Model. Softw. 2012, 37, 134–135. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Aliferis, C.F. Towards Principled Feature Selection: Relevancy, Filters and Wrappers. In International Workshop on Artificial Intelligence and Statistics; Christopher, M.B., Brendan, J.F., Eds.; PMLR, Proceedings of Machine Learning Research: Philadelphia, PA, USA, 2003; Volume R4, pp. 300–307. [Google Scholar]
- Yu, K.; Guo, X.; Liu, L.; Li, J.; Wang, H.; Ling, Z.; Wu, X. Causality-based feature selection: Methods and evaluations. ACM Comput. Surv. 2020, 53, 1–36. [Google Scholar] [CrossRef]
- Ling, Z.; Yu, K.; Zhang, Y.; Liu, L.; Li, J. Causal learner: A toolbox for causal structure and markov blanket learning. Pattern Recognit. Lett. 2022, 163, 92–95. [Google Scholar] [CrossRef]
- Schoonjans, F.; Zalata, A.; Depuydt, C.; Comhaire, F. MedCalc: A new computer program for medical statistics. Comput. Methods Programs Biomed. 1995, 48, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using ‘ggplot2′, R Package version 0.3; R Core Team: Vienna, Austria, 2017; p. 1. [Google Scholar]
- Kramer, O.; Kramer, O. K-nearest neighbors. In Dimensionality Reduction with Unsupervised Nearest Neighbors; Springer: Berlin/Heidelberg, Germany, 2013; pp. 13–23. [Google Scholar]
- Leung, K.M. Naive bayesian classifier. Polytech. Univ. Dep. Comput. Sci./Financ. Risk Eng. 2007, 2007, 123–156. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Meyer, P.E.; Bontempi, G. On the use of variable complementarity for feature selection in cancer classification. In Proceedings of the Applications of Evolutionary Computing: EvoWorkshops 2006: EvoBIO, EvoCOMNET, EvoHOT, EvoIASP, EvoINTERACTION, EvoMUSART, and EvoSTOC, Budapest, Hungary, 10–12 April 2006; Springer: Berlin/Heidelberg, Germany, 2006; pp. 91–102. [Google Scholar]
- Kumari, P.; Kumar, S.; Sethy, M.; Bhue, S.; Mohanta, B.K.; Dixit, A. Identification of therapeutically potential targets and their ligands for the treatment of OSCC. Front. Oncol. 2022, 12, 910494. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Zeng, Y.; Huang, D. Feature selection approaches identify potential plasma metabolites in postmenopausal osteoporosis patients. Metabolomics 2022, 18, 86. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Ru, X.; Sun, P.; Wang, J. A hybrid feature selection algorithm and its application in bioinformatics. PeerJ Comput. Sci. 2022, 8, e933. [Google Scholar] [CrossRef]
- Gnana, D.A.A.; Balamurugan, S.A.A.; Leavline, E.J. Literature review on feature selection methods for high-dimensional data. Int. J. Comput. Appl. 2016, 136, 9–17. [Google Scholar]
- Llamedo, M.; Martínez, J.P. Heartbeat Classification Using Feature Selection Driven by Database Generalization Criteria. IEEE Trans. Biomed. Eng. 2011, 58, 616–625. [Google Scholar] [CrossRef]
- Koller, D.; Sahami, M. Toward Optimal Feature Selection; Stanford InfoLab: Stanford, CA, USA, 1996. [Google Scholar]
- Guo, B.; Nixon, M.S. Gait feature subset selection by mutual information. IEEE Trans. Syst. MAN Cybern.-Part A Syst. Hum. 2008, 39, 36–46. [Google Scholar]
- Ircio, J.; Lojo, A.; Mori, U.; Lozano, J.A. Mutual information based feature subset selection in multivariate time series classification. Pattern Recognit. 2020, 108, 107525. [Google Scholar] [CrossRef]
- Walsh, A.M.; Lazzara, M.J. Regulation of EGFR trafficking and cell signaling by Sprouty2 and MIG6 in lung cancer cells. J. Cell Sci. 2013, 126, 4339–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbols | Description | Symbols | Description |
|---|---|---|---|
| Dataset | The sample size of | ||
| Original feature set | The feature size of | ||
| Class labels | The feature size of | ||
| The selected feature subset | The size of | ||
| Optimal feature subset | Feature number | ||
| Feature number | The candidate feature set | ||
| The candidate feature | The objective function |
| No. | Dataset | CONDRED | MIM | mRMR | DISR | CIFE | WBFS |
|---|---|---|---|---|---|---|---|
| 1 | Lung | 46.43 ± 16.94 * | 77.68 ± 18.08 * | 85.71 ± 15.06 | 86.07 ± 14.72 | 61.25 ± 16.79 * | 88.93 ± 8.94 |
| 2 | Breast | 93.32 ± 3.07 * | 95.61 ± 2.77 | 95.08 ± 1.61 | 93.33 ± 3.94 * | 92.79 ± 2.67 * | 95.43 ± 2.06 |
| 3 | Colon | 83.81 ± 13.65 | 87.14 ± 13.05 | 90.24 ± 11.53 v | 83.81 ± 13.65 | 87.14 ± 6.85 | 85.48 ± 12.27 |
| 4 | Ionosphere | 85.46 ± 5.14 | 86.32 ± 4.24 | 85.44 ± 4.97 | 86.32 ± 5.02 v | 85.17 ± 6.17 | 84.60 ± 5.11 |
| 5 | Isolet | 20.06 ± 2.74 * | 29.74 ± 3.50 * | 65.06 ± 4.29 v | 57.69 ± 3.32 * | 50.90 ± 4.00 * | 59.94 ± 3.45 |
| 6 | Krvskp | 88.95 ± 1.69 * | 96.25 ± 0.94 | 94.15 ± 1.26 * | 93.37 ± 1.04 * | 94.4 ± 1.27 * | 96.25 ± 0.94 |
| 7 | Landsat | 84.51 ± 1.54 | 83.03 ± 1.12 * | 84.79 ± 1.10 | 84.16 ± 1.64 | 85.30 ± 1.16 | 84.48 ± 1.76 |
| 8 | Madelon | 76.38 ± 2.43 * | 76.85 ± 2.18 * | 57.5 ± 3.82 * | 81.42 ± 1.78 * | 81.65 ± 3.07 | 82.92 ± 1.84 |
| 9 | Musk | 50.85 ± 2.18 * | 87.06 ± 1.00 * | 83.13 ± 0.95 * | 69.25 ± 1.82 * | 76.66 ± 1.82 * | 91.66 ± 1.37 |
| 10 | Sonar | 75.45 ± 9.19 | 83.17 ± 10.51 | 78.33 ± 8.59 | 83.64 ± 9.12 | 80.24 ± 8.99 | 81.17 ± 10.29 |
| 11 | Splice | 82.87 ± 1.78 | 82.43 ± 2.02 | 82.43 ± 2.02 | 82.43 ± 2.02 | 79.97 ± 2.47 * | 82.43 ± 2.02 |
| 12 | Waveform | 80.52 ± 1.44 | 80.52 ± 1.44 | 80.52 ± 1.44 | 80.52 ± 1.44 | 69.94 ± 2.02 * | 80.52 ± 1.44 |
| Average | 72.38 | 80.48 | 81.87 | 81.83 | 78.78 | 84.48 | |
| W/T/L | 6/6/0 | 5/7/0 | 3/7/2 | 5/6/1 | 7/5/0 | ||
| No. | Dataset | CONDRED | MIM | mRMR | DISR | CIFE | WBFS |
|---|---|---|---|---|---|---|---|
| 1 | Lung | 51.96 ± 24.43 | 58.75 ± 24.69 | 61.61 ± 23.35 | 61.61 ± 23.35 | 60.18 ± 22.59 | 60.18 ± 22.11 |
| 2 | Breast | 89.99 ± 3.60 * | 92.79 ± 3.14 | 93.32 ± 2.84 | 62.74 ± 6.29 * | 92.79 ± 2.26 | 93.32 ± 2.96 |
| 3 | Colon | 80.48 ± 10.75 * | 88.57 ± 13.65 | 88.57 ± 13.65 | 88.57 ± 13.65 | 69.05 ± 18.58 * | 88.81 ± 11.06 |
| 4 | Ionosphere | 72.66 ± 9.70 * | 64.10 ± 6.04 * | 64.10 ± 6.04 * | 64.10 ± 6.04 * | 80.35 ± 10.45 * | 84.62 ± 7.75 |
| 5 | Isolet | 22.24 ± 3.96 * | 24.17 ± 2.99 * | 26.92 ± 4.46 * | 16.79 ± 2.80 * | 55.83 ± 3.29 v | 43.08 ± 4.35 |
| 6 | Krvskp | 52.22 ± 1.88 | 52.22 ± 1.88 | 52.22 ± 1.88 | 52.22 ± 1.88 | 52.22 ± 1.88 | 52.22 ± 1.88 |
| 7 | Landsat | 75.46 ± 1.71 * | 72.82 ± 1.76 * | 76.53 ± 1.85 | 76.64 ± 1.52 | 77.62 ± 2.16 v | 76.38 ± 1.94 |
| 8 | Madelon | 59.58 ± 3.10 | 59.62 ± 2.36 v | 58.96 ± 2.31 | 59.19 ± 2.52 | 59.15 ± 3.65 | 59.12 ± 2.15 |
| 9 | Musk | 73.78 ± 1.49 * | 77.39 ± 1.79 * | 61.56 ± 1.60 * | 82.69 ± 0.96 v | 88.88 ± 0.93 v | 78.87 ± 1.35 |
| 10 | Sonar | 64.79 ± 13.19 * | 70.12 ± 15.74 | 66.74 ± 16.06 | 68.67 ± 15.47 * | 71.57 ± 9.61 | 73.48 ± 16.34 |
| 11 | Splice | 88.69 ± 8.38 | 88.62 ± 8.37 | 88.62 ± 8.37 | 88.62 ± 8.37 | 87.74 ± 7.96 * | 88.62 ± 8.37 |
| 12 | Waveform | 79.08 ± 1.26 | 79.08 ± 1.26 | 79.08 ± 1.26 | 79.08 ± 1.26 | 77.34 ± 0.91 * | 79.08 ± 1.26 |
| Average | 67.58 | 69.02 | 68.19 | 66.74 | 72.73 | 73.15 | |
| W/T/L | 7/5/0 | 4/7/1 | 3/9/0 | 4/7/1 | 4/5/3 | ||
| No. | Dataset | CONDRED | MIM | mRMR | DISR | CIFE | WBFS |
|---|---|---|---|---|---|---|---|
| 1 | Lung | 43.75 ± 24.12 | 46.25 ± 24.69 * | 41.25 ± 27.36 | 46.25 ± 24.69 v | 28.93 ± 23.28 * | 42.5 ± 24.09 |
| 2 | Breast | 93.32 ± 2.60 * | 94.38 ± 2.72 | 95.25 ± 1.90 | 92.97 ± 2.35 * | 92.79 ± 3.25 * | 95.42 ± 2.40 |
| 3 | Colon | 62.86 ± 22.14 | 69.29 ± 19.97 v | 64.52 ± 23.07 | 64.52 ± 23.07 | 64.52 ± 23.07 | 64.52 ± 23.07 |
| 4 | Ionosphere | 83.47 ± 6.99 * | 88.60 ± 4.48 | 89.16 ± 4.24 v | 88.90 ± 4.72 * | 87.46 ± 4.71 | 87.18 ± 5.25 |
| 5 | Isolet | 28.33 ± 2.36 * | 33.78 ± 4.34 * | 68.21 ± 2.92 v | 61.15 ± 1.87 * | 61.79 ± 2.59 * | 64.68 ± 3.46 |
| 6 | Krvskp | 75.28 ± 2.55 * | 77.63 ± 2.10 | 72.37 ± 2.39 * | 72.03 ± 2.38 * | 72.59 ± 2.03 * | 77.63 ± 2.10 |
| 7 | Landsat | 82.77 ± 1.40 | 80.57 ± 1.56 * | 82.78 ± 1.40 | 82.81 ± 1.62 | 83.37 ± 1.35 v | 82.69 ± 1.50 |
| 8 | Madelon | 65.85 ± 1.41 * | 65.62 ± 1.28 * | 60.35 ± 1.57 * | 67.77 ± 1.94 * | 67.81 ± 2.56 * | 69.50 ± 2.39 |
| 9 | Musk | 84.59 ± 1.27 * | 84.60 ± 1.23 * | 84.59 ± 1.27 * | 84.59 ± 1.27 * | 90.21 ± 0.93 * | 91.12 ± 1.10 |
| 10 | Sonar | 64.83 ± 8.47 * | 74.00 ± 12.71 * | 76.4 ± 13.56 | 74.95 ± 12.35 | 75.40 ± 10.31 | 76.88 ± 11.64 |
| 11 | Splice | 89.39 ± 2.23 | 89.17 ± 2.07 | 89.17 ± 2.07 | 89.17 ± 2.07 | 87.62 ± 1.78 * | 89.17 ± 2.07 |
| 12 | Waveform | 83.62 ± 1.97 | 83.62 ± 1.97 | 83.62 ± 1.97 | 83.62 ± 1.97 | 77.32 ± 1.10 * | 83.62 ± 1.97 |
| Average | 72.38 | 80.48 | 81.87 | 81.83 | 78.78 | 83.01 | |
| W/T/L | 7/5/0 | 6/5/1 | 3/7/2 | 6/5/1 | 8/3/1 | ||
| No. | Protein Biomarker | AUC | Sensitivity | Specificity | p-Value |
|---|---|---|---|---|---|
| 1 | BRD4 | 0.749 | 59.38 | 82.60 | <0.0001 |
| 2 | CD26 | 0.824 | 75.70 | 75.40 | <0.0001 |
| 3 | DUSP4 | 0.733 | 67.40 | 67.40 | <0.0001 |
| 4 | GAPDH | 0.775 | 73.50 | 67.70 | <0.0001 |
| 5 | GSK3ALPHABETA | 0.765 | 66.50 | 75.40 | <0.0001 |
| 6 | IGFBP2 | 0.722 | 65.20 | 71.80 | <0.0001 |
| 7 | INPP4B | 0.767 | 74.20 | 66.90 | <0.0001 |
| 8 | MIG6 | 0.833 | 80.00 | 72.70 | <0.0001 |
| 9 | NDRG1_pT346 | 0.729 | 62.20 | 76.50 | <0.0001 |
| 10 | TFRC | 0.876 | 81.20 | 80.90 | <0.0001 |
| 11 | The combination of eight biomarkers | 0.960 | 90.15 | 92.54 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Gao, X.; Ru, X.; Sun, P.; Wang, J. The Weight-Based Feature Selection (WBFS) Algorithm Classifies Lung Cancer Subtypes Using Proteomic Data. Entropy 2023, 25, 1003. https://doi.org/10.3390/e25071003
Wang Y, Gao X, Ru X, Sun P, Wang J. The Weight-Based Feature Selection (WBFS) Algorithm Classifies Lung Cancer Subtypes Using Proteomic Data. Entropy. 2023; 25(7):1003. https://doi.org/10.3390/e25071003
Chicago/Turabian StyleWang, Yangyang, Xiaoguang Gao, Xinxin Ru, Pengzhan Sun, and Jihan Wang. 2023. "The Weight-Based Feature Selection (WBFS) Algorithm Classifies Lung Cancer Subtypes Using Proteomic Data" Entropy 25, no. 7: 1003. https://doi.org/10.3390/e25071003
APA StyleWang, Y., Gao, X., Ru, X., Sun, P., & Wang, J. (2023). The Weight-Based Feature Selection (WBFS) Algorithm Classifies Lung Cancer Subtypes Using Proteomic Data. Entropy, 25(7), 1003. https://doi.org/10.3390/e25071003