Experimental
General
Melting points were determined by the capillary tube method, and are uncorrected. IR spectra (KBr pellets) were recorded on a Bio-Rad FTS 135 spectrophotometer. Optical rotations were measured on a WZZ-3 digital polarimeter with a sodium lamp (λ=589nm, D line). 1H- and 13C‑NMR spectra were recorded on a Bruker AV400 instrument. Chemical shifts (δ) are reported in ppm relative to TMS used as internal standard. Mass spectra were recorded on a LCQ analytical Advantage MAX spectrometer. Organic solvents were dried by standard methods when necessary. Thin-layer chromatography (TLC) was performed on glass sheets coated with silica gel (type GF254, Qingdao Haiyang). Flash column chromatography was carried out using 200-300 mesh silica gel.
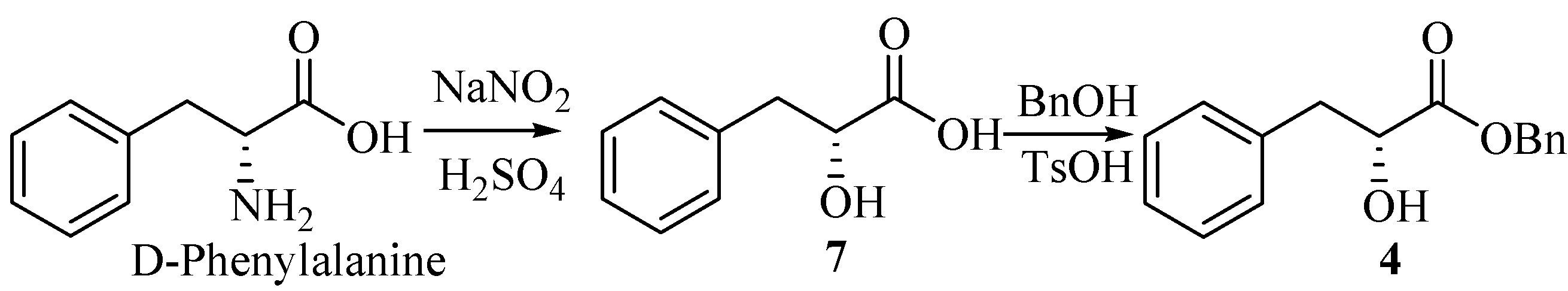
(R)-2-hydroxy-3-phenylpropanoic acid (7).
A solution of 40% sodium nitrite (12 mL) was added into a solution of D-phenylalanine (6.60 g, 40 mmol) in H2SO4 (2 mol/L, 45 mL) over 2 h under ice cooling. The mixture was stirred for 6h at 0°C, and then at room temperature for 12 h. Additional H2SO4 (2 mol/L, 10 mL) and sodium nitrite (40%, 6 mL) were added as above. The reaction mixture was saturated by NaCl, followed by extraction with ethyl acetate (3×60 mL). The combined extracts were washed with brine (200 mL), dried (MgSO4), filtered and evaporated to give a white solid which upon recrystallization from diethyl ether, gave the product 7 (6.54g, 98.5%), m.p. 125-127°C (lit.[2], 123-124°C), =+38.6°(c 1.00, DMF) [lit.[2], =+26.7°(c 3.80, acetone)]
(R)-benzyl-2-hydroxy-3-phenylpropanoate (4).
Into a 100-mL round-bottom flask equipped with a Stark and Dean distilling receiver were placed compound 7 (1.66 g, 10 mmol), p-toluenesulfonic acid monohydrate (0.02 g, 0.12 mmol), benzyl alcohol (1.6 mL, 15 mmol), and toluene (30 mL). Then the mixture was heated under reflux. When water was no longer distilled off (about 4 h), the reaction mixture was permitted to cool to room temperature. Then ether (30 mL) and saturated aqueous Na2CO3 (50 mL) were added. The two layers were separated, and the water layer was extracted with ether (2×30mL). The combined extracts were washed with brine (100 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 10:1) to give 4 (1.45g, 56.6%). =+54.9°(c 1.5, CH2Cl2) [lit.[2], =+55.2°(c 1.88, CH2Cl2)]; IR (KBr): 3484, 3034, 2928, 1737, 1495, 1450, 1380, 1264, 1189, 1094, 745, 698 cm-1; 1H-NMR (400 MHz, CDCl3) δ: 7.18~7.41 (m, 10H), 5.20 (s, 2H), 4.51 (dd, 1H, J1=4.8 Hz, J2=6.4 Hz), 3.15 (dd, 1H, J1=4.8 Hz, J2=13.6 Hz), 3.01 (dd, 1H, J1=6.4 Hz, J2=13.6 Hz), 2.92 (br, OH); 13C-NMR (100 MHz, CDCl3) δ: 173.85, 136.09, 134.94, 129.40, 128.50, 128.44, 128.23, 126.67, 71.17, 67.18, 40.34.
(tert-Butoxycarbonylmethylamino)acetic acid (5).
To a cooled (0°C) solution of Boc-Gly-OH 8 (7.33 g, 41.9 mmol) and MeI (10.6 mL, 170 mmol) in dry THF (200 mL), was added NaH (5.23g, 120 mmol, 55% in mineral oil) cautiously and the mixture was with stirred gently overnight. CH3I (5.3 mL) and NaH (1.75 g) were added in again by the above procedure to push the reaction to completion. Ethyl acetate (100 mL) was then added, followed by water (15 mL) added dropwise to destroy the excess NaH. The solution was evaporated to dryness, and the oily residue partitioned between ether (60 mL) and water (100 mL). The ether layer was washed with saturated aqueous NaHCO3 (50 mL), and the combined aqueous extracts acidified to pH 2 with 1 mol/L HCl under ice cooling. The product was extract into ethyl acetate (2×100 mL). Then the extracts were washed with water (100 mL), 5% aqueous sodium thiosulfate (2×100 mL, to remove iodine), and brine (100 mL), dried (MgSO4), filtered, and evaporated to give a pale white oil 5 (7.32g, 92.5%).
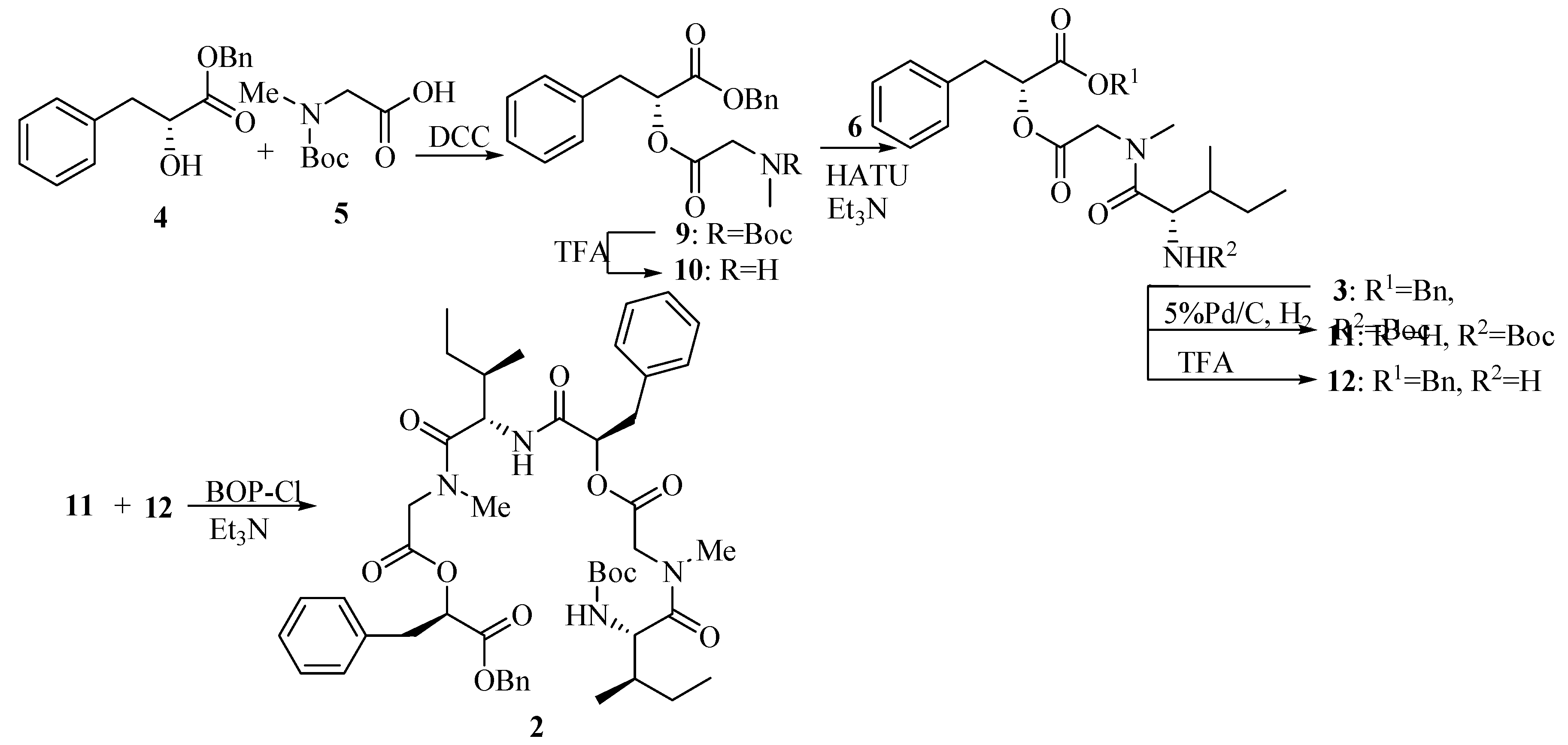
2-[2-(tert-Butoxycarbonylmethylamino)acetoxy]-3-phenylpropionic acid benzyl ester (9).
Compound 4 (1.29 g, 5.0 mmol) was dissolved in dry CH2Cl2 (10 mL) and cooled to 0°C. A solution of N-Me-Boc-Gly-OH 5 (0.95 g, 5.0 mmol) in CH2Cl2 (10 mL) was added in one portion followed by HOBt (0.73 g, 5.4 mmol) and DMAP (0.66 g, 5.4 mmol) and the resulting suspension was then stirred for 15 min. A solution of DCC (1.12 g, 5.4 mmol) in dry CH2Cl2 (10 mL) was added to the suspension over 10 min and the mixture was slowly warmed to room temperature and stirred for 24 h. The mixture was evaporated in vacuo to leave a solid and then taken up in ethyl acetate (50 mL), filtered to remove DCU (by product). The filtrate was washed with saturated aqueous NaHCO3 (2×50 mL) and brine (50 mL). The organic layer was dried (MgSO4) and evaporated in vacuo to leave a residue and then purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 10:1) to yield title compound 9 (1.56 g, 72.5%). =+16.3°(c 1.0, CHCl3); IR (KBr): 2970, 2932, 1754, 1703, 1453, 1381, 1243, 1179, 1160, 1070, 746, 703cm-1; 1H-NMR (400 MHz, CDCl3) δ (rotamer): 7.14~7.32 (m, 10H), 5.30~5.32 (m, 1H), 5.08~5.15 (m, 2H), 3.91~4.05 (m, 2H), 3.10~3.18 (m, 2H), 2.79, 2.78 (s, 3H, rotamer), 1.26~1.46(m, 9H); 13C-NMR (100 MHz, CDCl3) δ (rotamer): 169.05, 168.64, 155.60, 154.95, 135.20, 135.13, 134.84, 129.16, 129.06, 128.31, 128.28, 128.26, 128.19, 128.09, 128.05, 126.81, 79.83, 79.77, 73.09, 73.01, 66.86, 50.39, 49.68, 36.98, 34.93, 28.06, 27.87.
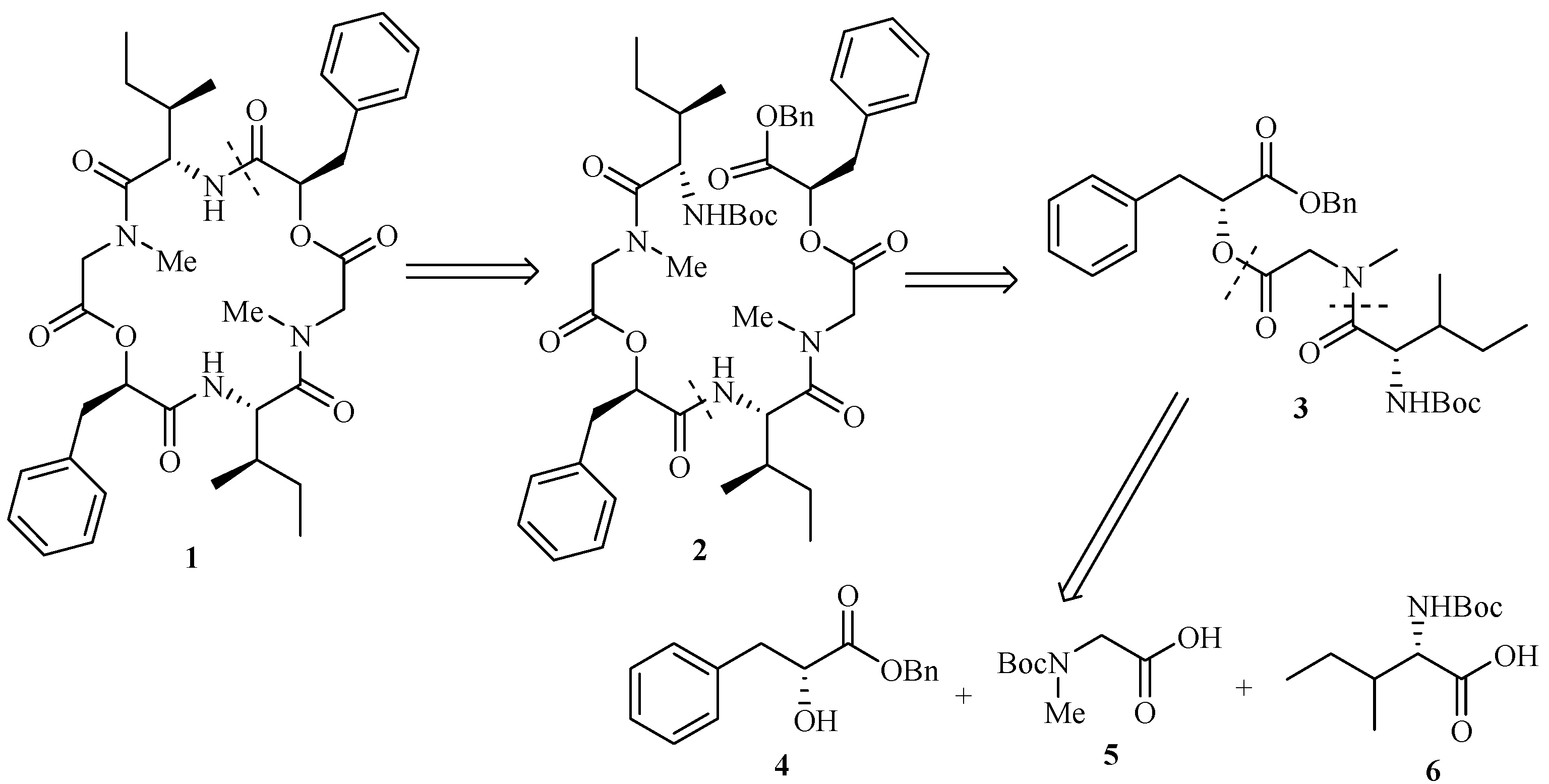
2-{2-[(2-tert-Butoxycarbonylamino-3-methylpentanoyl)methylamino]acetoxy}-3-phenylpropionic acid benzyl ester (3).
To a solution of 9 (1.70 g, 4.0 mmol) in dry CH2Cl2 (6 mL) was added TFA (2 mL, 26.0 mmol) dropwise at 0°C with stirring. The mixture then was slowly warmed to room temperature and stirred for 5 h. The reaction mixture was concentrated, the residue dried under high vacuum and dissolved in dry CH2Cl2 (10 mL). A solution of N-Boc-Ile-OH 6 (1.01 g, 4.4 mmol) in dry CH2Cl2 (10 mL) was added in one portion followed by HATU (99%) (1.84 g, 4.8 mmol). Triethylamine (5.6 mL, 39.8 mmol) was added to the mixture dropwise under ice cooling. Then the mixture was warmed to room temperature slowly and stirred for 12 h. The reaction was quenched by adding saturated aqueous NH4Cl (2mL) dropwise and evaporated in vacuo followed by dissolving in ethyl acetate (150 mL). The mixture was washed with saturated aqueous NaHCO3 (2×100 mL) and brine (100 mL), dried (MgSO4), filtered and evaporated in vacuo to leave a residue which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 5:1) to afford tridepsipeptide 3 (1.74 g, 80.9%). =+6.3° (c 1.0 CHCl3); IR (KBr): 3409, 3316, 2969, 1753, 1708, 1652, 1497, 1402, 1377, 1246, 1176, 1075, 1031, 745, 703cm-1; 1H-NMR (400 MHz, CDCl3) δ (rotamer): 7.10~7.34 (m, 10H), 5.31 (dd, 1H, J1=5.2 Hz, J2=7.6 Hz), 5.04~5.17 (m, 3H), 4.51 (br, NH), 4.53 (d, 1H, J=17.6 Hz), 3.80 (d, 1H, J=17.6 Hz), 3.16(dd, 1H, J1=5.2 Hz, J2=14.4 Hz), 3.10 (dd, 1H, J1=7.6 Hz, J2=14.4 Hz), 3.00 (s, 3H), 1.66~1.71 (m, 1H), 1.40 (s, 9H), 0.88~0.96 (m, 8H); 13C-NMR (100 MHz, CDCl3) δ (rotamer): 173.12, 168.95, 168.38, 155.83, 135.23, 134.97, 129.33, 128.56, 128.52, 128.49, 128.42, 128.36, 127.13, 79.48, 73.43, 67.23, 54.22, 49.16, 38.03, 37.15, 36.36, 28.32, 23.91, 15.55, 11.29.
2-(2-{[2-(2-{2-[(2-tert-Butoxycarbonylamino-3-methylpentanoyl)methylamino]acetoxy}-3-phenyl- propionylamino)-3-methylpentanoyl]methylamino}acetoxy)-3-phenylpropionic acid benzyl ester (2).
A solution of 3 (0.26 g, 0.5 mmol) in ethyl acetate (30 mL) was treated with Pd-C (5%, 0.52 g), and the black suspension was stirred at room temperature under an atmosphere of H2 for 12 h. The catalyst was removed by filtration, and the filtrate was concentrated in vacuo to give crude 11 (used in the next step without further purification). To a solution of 3 (0.20 g, 0.4 mmol) in dry CH2Cl2 (2 mL) was added TFA (0.5 mL, 6.5 mmol) dropwise at 0°C with stirring. The mixture then was slowly warmed to room temperature and stirred for 5 h. The reaction mixture was concentrated, and the residue dried under high vacuum to give crude 12 (used directly in the next step). TFA salt 12 was dissolved in dry CH2Cl2 (5 mL). A solution of 11 in dry CH2Cl2 (5 mL) was added in one portion with stirring and cooled to 0°C. BOP-Cl (0.13 g, 0.5 mmol) was added at 0°C followed by triethylamine (0.5 mL, 3.7 mmol). The mixture was then warmed to room temperature and stirred for 12 h. The reaction was quenched by adding saturated aqueous NH4Cl (2 mL) dropwise and evaporated in vacuo followed by dissolving in ethyl acetate (50 mL). The mixture was washed with saturated aqueous NaHCO3 (2×50 mL) and brine (50 mL), dried (MgSO4), filtered and evaporated in vacuo to leave a residue which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 2:1) to give hexadepsipeptide 2 (0.16g, 76.6%).
=+1.06º(c 3.16, CHCl3); IR (KBr): 3309, 2963, 2931, 1753, 1705, 1645, 1501, 1465, 1406, 1376, 1250, 1176, 1071, 745, 707cm-1; 13C-NMR (100 MHz, CDCl3) δ: 174.24, 172.53, 168.84, 168.67, 168.19, 167.47, 156.06, 136.29, 135.28, 134.90, 130.83, 129.32, 129.29, 129.25, 128.56, 128.53, 128.48, 128.45, 128.40, 128.39, 128.35, 128.28, 127.07, 126.90, 126.79, 79.19, 74.79, 73.42, 67.21, 65.47, 54.00, 52.38, 49.06, 38.00, 37.27, 36.83, 36.28, 30.51, 29.61, 28.40-28.38 (3C), 24.27, 24.24, 15.37, 15.20, 10.84, 10.57; MS: m/z= 895.5 (M+Na)+; Anal. Calcd for C48H64N4O11: C, 66.04; H, 7.39; N, 6.42; O, 20.16. Found: C, 66.18; H, 7.36; N, 6.40; O, 20.06.

{kind=link}
{kind=link}
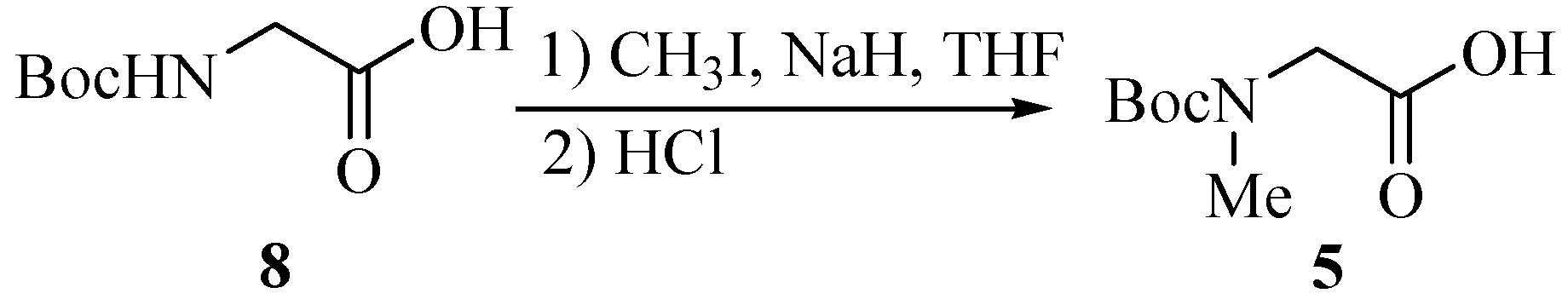{kind=link}
{kind=link}



