General
Melting points were determined on a Fisher-Johns melting point apparatus and are uncorrected. (IR) spectra were taken on a Perkin Elmer FT-IR 1600 spectrometer. 1H- (200 MHz) and 13C-NMR (75 MHz) spectra were recorded on a Bruker Avance DPX-300 MHz spectrometer. Spectra were run in CDCl3 with tetramethylsilane (TMS) used as internal standard. The EIMS data was obtained on a Finnigan MAT-90 instrument and all experiments were performed in the electron-impact mode (EI) at 70 eV using a direct insertion probe.
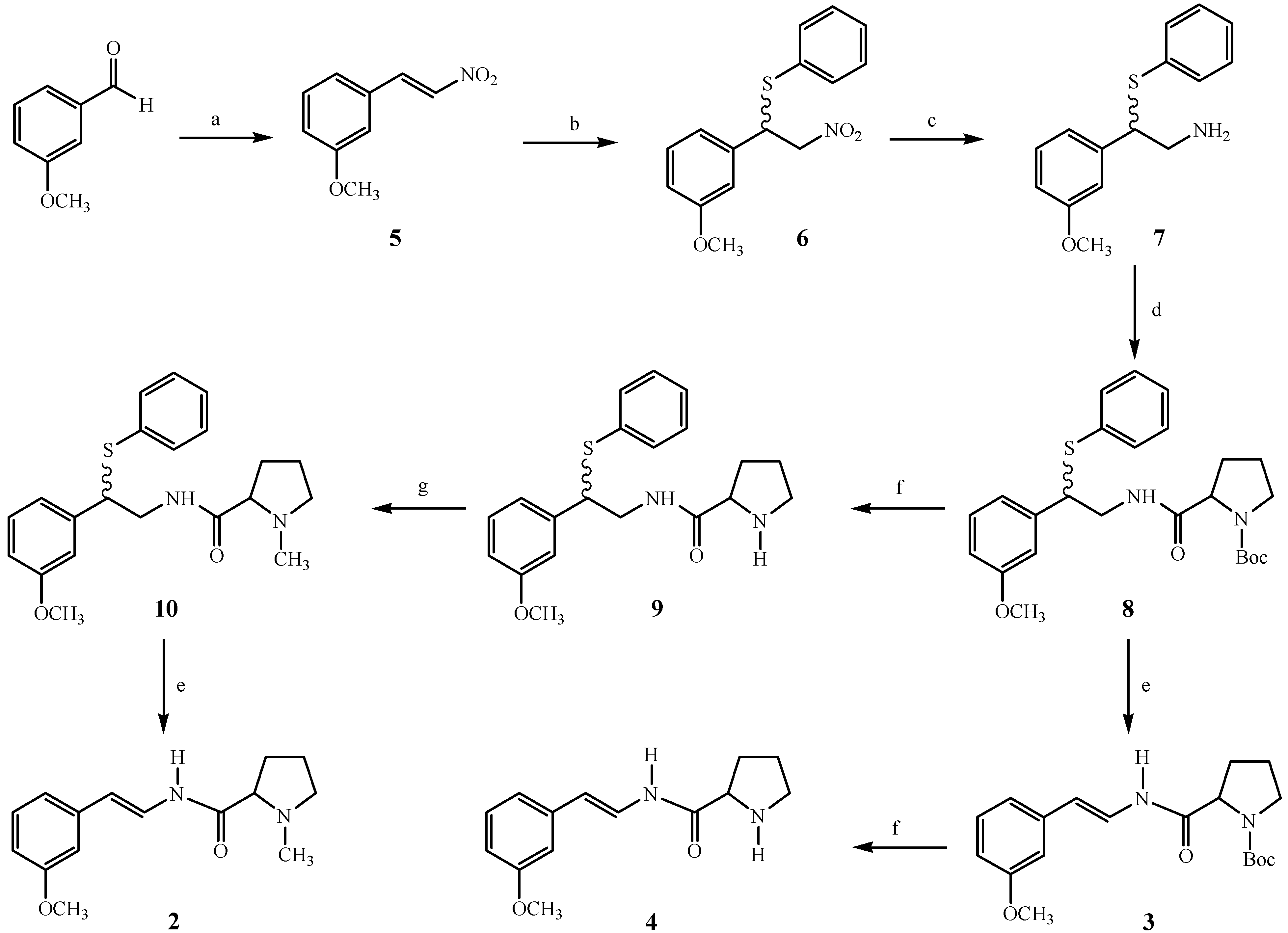
(E)-3-Methoxy-β-nitrostyrene (5): To a solution of m-anisaldehyde (73.4 mmol, 10.0 g) in glacial AcOH (120 mL) was added AcONH4 (2.71 g) and nitromethane (13.1 mL). The solution was heated under reflux for 1 h. The mixture was cooled and the precipitate was washed with water and recrystalized from AcOH/H2O to give a yellowish solid (10.6 g, 81%); mp 88-89° C; IR (KBr): 3108, 1636, 1577, 1511 cm-1; 1H-NMR: δ 7.97 (d, J = 14.0 Hz), 7.56 (d, J = 14.0 Hz), 7.41-7.02 (m, 4H), 3.85 (s, 3H, OCH3).
2-(3-Methoxyphenyl)-2-(thiophenyl)-1-nitroethane (6): Compound 5 (22.3 mmol, 4.0 g) was dissolved in CH2Cl2 (100 mL) and was added thiophenol (22.9 mmol, 2.6 mL) and 0.5 mL of N-isopropylcyclo-hexylamine. The resulting solution was stirred for 1 h. at rt. The solution was concentrated and subjected to flash chromatography using hexane/CH2Cl2 (80:20). Removal of the solvent gave a brownish oil (5.8 g, 90%). IR (film): 3057, 2957, 1599, 1554, 1262 cm-1; 1H-NMR: δ 7.45-7.22 (m, 6H), 6.85-6.79 (m, 3H), 4.83-4.60 (m, 3H), 3.75 (s, 3H, OCH3).
2-(3-Methoxyphenyl)-2-(thiophenyl)-1-aminoethane (7): Compound 6 (17.3 mmol, 5.0 g) was dissolved in AcOH (46 mL) and Zn powder (172.4 mmol, 11.3 g) was added. Then conc. HCl (37 mL) was added dropwise and the solution was stirred overnight at rt. The solution was neutralized with 2 N NH4OH. The residue was extracted into ethyl acetate (2 × 100 mL) and the solvent was removed under reduced pressure to give a yellowish green oil (2.0 g, 45%); IR (film): 3372, 3057, 2936, 1587, 1483, 1261, 1044, 694 cm-1; 1H-NMR: δ 7.4-7.18 (m, 6H), 6.96-6.70 (m, 3H), 4.12 (t, J = 8 Hz), 3.77 (s, 3H, OCH3), 3.20-3.00 (m, 2H), 2.00-1.40 (bs, 2H, NH2).
2(S)-N-[2(3-Methoxyphenyl)-2-(thiophenyl)ethanyl]-1-t-butoxycarbonylproline (8): To a solution of (S)-N-t-butoxycarbonylproline (19.5 mmol, 4.1 g) in dry THF (100 mL) was added dicyclohexyl-carbodiimide (19.5 mmol, 4.0 g), 1-hydroxybenzotriazole (19.5 mmol, 2.6 g) and 4-N,N-dimethyl-aminopyridine in catalytic amounts. A solution of 7 (19.5 mmol, 5.05 g) in dry THF (20 mL) was added and the suspension was stirred for 24 h, filtered and the solvent was removed in vacuo. The residue was purified by flash chromatography using CH2Cl2/MeOH (95:5). Removal of the solvent gave a brownish oil (1.8 g, 20%); IR (film): 3313, 3056, 2974, 1697 cm-1; 1H-NMR: δ 7.50-7.20 (m, 6H), 6.90-6.76 (m, 3H), 4.50-4.30 (m, 1H), 4.30-4.10 (m, 1H), 3.75 (s, 3H, OCH3), 3.90-3.50 (m, 2H), 3.50-3.20 (m, 2H), 2.40-1.60 (m, 4H), 1.39 (s, 9H, boc); 13C-NMR: δ 159.6, 140.7, 133.7, 132.2, 129.5, 128.8, 127.3, 127.2, 120.0, 113.3, 113.2, 113.1, 80.3, 55.1, 52.3, 46.9, 43.8, 43.6, 28.3: IEMS, m/z: M+ 456 (nd), [M-PhSH]+ 346 (23), 198 (3), 149 (62), 132 (2), 70 (100), 57 (33).
2(S)-N-[2(3-Methoxyphenyl)-2-(thiophenyl)ethanyl]-2-pyrrolinecarboxamide (9): Compound 8 (4.38 mmol, 2.0 g) was dissolved in acetonitrile (30 mL) and HF (50%, 0.7 mL) was added and the mixture heated overnight under reflux with magnetic stirring. The mixture was cooled and the solvent was removed in vacuo. The residue was treated with K2CO3 (50%, 20 mL) and extracted with dichloromethane (2 × 20 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to give 9 (1.51 g, 97%). Compound 9 was used in the next step without further purification. IR (film): 3322, 3054, 2960, 1665 cm-1.
2(S)-N-[2(3-Methoxyphenyl)-2-(thiophenyl)ethanyl]-1-methyl-2-pyrrolinecarboxamide (10): 9 (4.24 mmol, 1.5 g) was dissolved in acetonitrile (14 mL) and formaldehyde (37%, 2.3 mL) and sodium cyanoborohydride (9.19 mmol, 0.6 g) were added and the solution was stirred for 2 h. at rt. The solution was neutralized with AcOH and stirring was continued for 2 h. The solvent was removed under vacuum and the residue was washed with water and extracted with CH2Cl2 (2 × 15 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to give a brownish oil of the title compound as a mixture of diastereomers (1.2 g, 80%). IR (film): 3328, 3056, 2942, 1663, 1261 cm-1; 1H-NMR: δ 7.56 (bs, 2H, N-H), 7.38-7.160 (m, 12H), 6.88-6.75 (m, 6H), 4.42-4.32 (m, 2H), 3.87-3.55 (m, 4H), 3.76 (s, 6H, OCH3), 3.00-2.93 (m, 2H), 2.80 (dd, J =10.0, 4.0 Hz, 2H), 2.32-2.00 (m, 4H), 2.22 (s, 3H, NCH3), 2.15 (s, 3H, NCH3), 1.50-1.80 (m, 6H); 13C-NMR: δ 174.9, 174.8, 159.8, 141.0, 141.0, 134.1, 132.1, 132.1, 129.9, 129.6, 129.1, 128.9, 127.5, 127.4, 127.3, 120.4, 120.3, 113.4, 113.4, 113.4, 113.3, 68.8, 56.6, 56.5, 55.2, 52.2, 43.6, 43.5, 41.6, 41.5, 31.0, 30.9, 24.22.
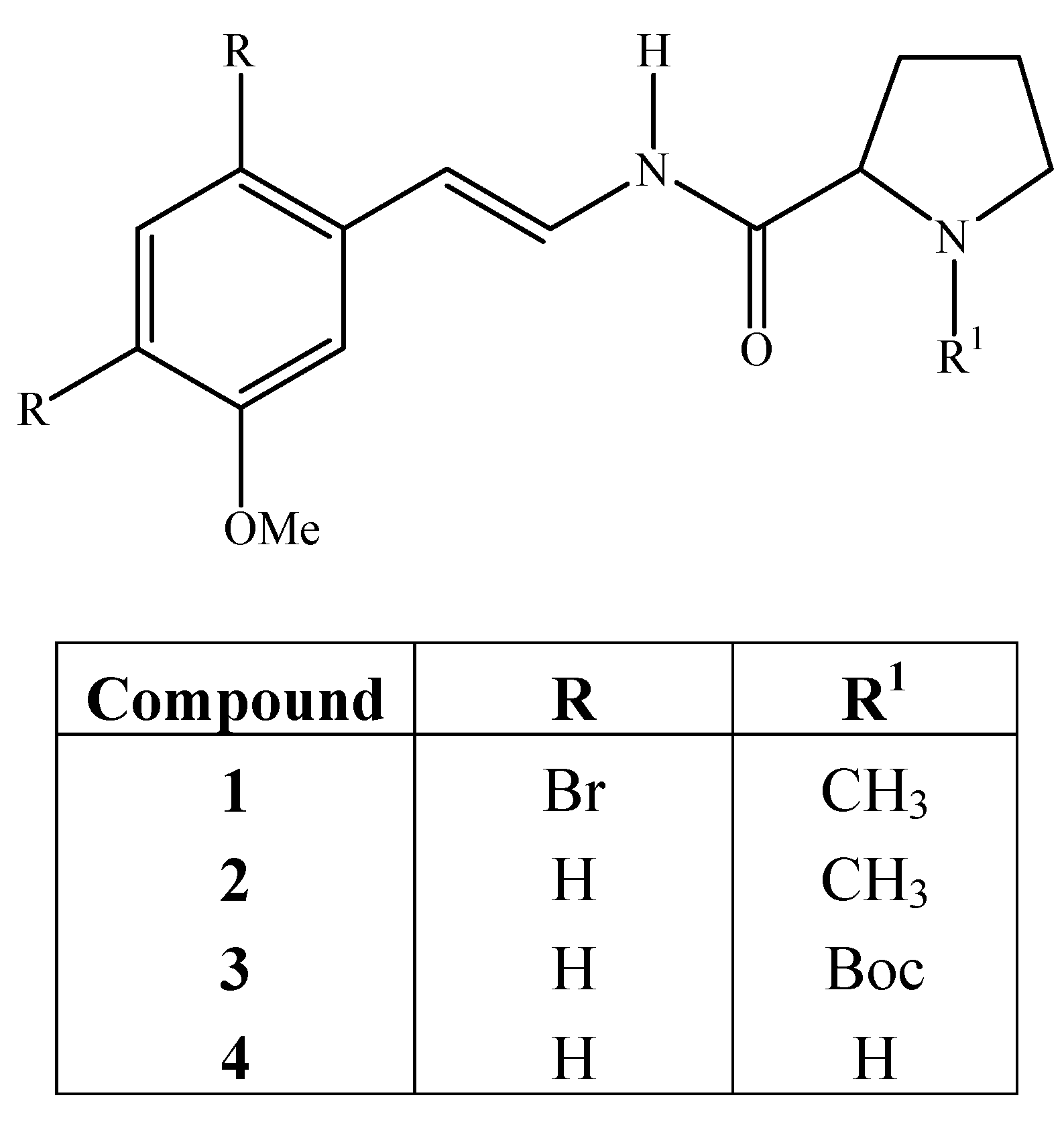
2(S)-N-[(E)-2(3-Methoxyphenyl)ethanyl]-1-methyl-2-pyrrolinecarboxamide (2): To a solution of 10 (1.7 mmol, 0.6 g) in methanol (10 mL) was added a solution of sodium periodate (1.9 mmol, 0.4 g) in water (10 mL) and the resulting mixture heated under reflux for 1.5 h. The methanol was removed in vacuo and the aqueous residue was extracted with dichloromethane (2 × 20 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to give sulfoxide as a brownish oil (0.57 g, 70%). The crude product was then used in the next step without purification. The sulfoxide (1.20 mmol, 0.465 g) was dissolved in toluene (30 mL), sodium carbonate was added (166 mg) and the mixture heated under reflux for 2 h. The solvent was removed under vacuum and the residue was purified by circular chromatography using CH2Cl2/MeOH (95:5). Removal of the solvent gave a brownish oil (0.16 g, 35%). IR (film): 3286, 2942, 1681, 1650, 1504, 1262 cm-1; 1H-NMR: δ 9.21 (d, J = 10.8 Hz, 1H, N-H), 7.40 (dd, J = 14.0, 12.0 Hz, 1H), 7.20 (t, J = 8.0 Hz, 1H), 6.94-6.85 (m, 2H), 6.73 (ddd, J = 8.2, 2.6, 0.8 Hz, 1H), 6.17 (d, J = 14.0 Hz, 1H), 3.81 (s, 3H, OCH3), 3.24-3.17 (m, 1H), 3.07 (dd, J = 9.8, 4.8 Hz, 1H), 2.42 (s, 3H, NCH3), 2.50-2.20 (m, 2H), 2.00-1.7 0 (m, 3H); 13C-NMR: δ 172.2, 159.9, 137.7, 129.6, 122.5, 118.4, 113.3, 112.5, 110.6, 68.7, 56.7, 55.2, 41.9, 31.0, 24.5; IEMS, m/z: M+ 260 (26), 132 (15), 112 (18), 70 (100).
2(S)-N-[(E)-2(3-Methoxyphenyl)ethanyl]-1-t-butoxycarbonyl-2-pyrrolinecarboxamide (3): Compound 3 was synthesized from 8 (2.2 mmol, 1.0 g) using the same procedure described for amide 2. Compound 3 was obtained as a white solid (0.30 g, 30%); IR (KBr): 3283, 2974, 1695, 1653, 1400, 1257, 1161, 774 cm-1; 1H-NMR: δ 9.28 (bs, 1H, N-H), 7.46 (dd, J = 14.6, 10.8 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 6.90 (d, J = 7.8, 1H), 6.85 (d, J = 2.4, 1H), 6.73 (dd, J = 8.2, 2.4 Hz, 1H), 6.08 (d, J = 14.6 Hz, 1H), 4.42-4.22 (m, 1H), 3.77 (s, 3H, OCH3), 3.56-3.2 (m, 2H), 2.60-1.80 (m, 4H), 1.49 (s, 9H); IEMS, m/z M+ 346 (23), 245 (1), 198 (2), 149 (42), 132 (6), 70 (100), 57 (33).
2(S)-N-[(E)-2(3-Methoxyphenyl)ethanyl]-2-pyrrolinecarboxamide (4): Compound 4 was obtained from 3 (0.6 mmol, 0.2 g) as a brownish oil (0.13 g, 90%) using the same procedure described for amide 9. IR (film) 3272, 3062, 2956, 1679, 1649, 1528, 1260, 1043, 692 cm-1; 1H-NMR: δ 9.58 (d, J = 10.6 Hz, 1H, N-H), 7.44 (dd, J = 15.0, 11.3 Hz, 1H), 7.19 (t, J = 8.0 Hz, 1H), 6.91 (d, J = 7.8, 1H), 6.87 (d, J = 2.0 Hz, 1H), 6.72 (ddd, J = 8.0, 2.4, 0.8 Hz, 1H), 6.16 (d, J = 15 Hz, 1H), 3.91-3.75 (m, 1H), 3.80 (s, 3H, OCH3), 3.10-2.90 (m, 2H), 2.65-2.3 (bs, 1H, NH), 2.26-2.15 (m, 1H), 2.06-1.90 (m, 1H), 1.81-1.71 (m, 2H); 13C-NMR: δ 172.9, 160.0, 138.0, 129.8, 122.6, 118.5, 113.5, 112.6, 110.7, 60.5, 55.4, 47.5, 30.9, 26.4; IEMS m/z M+ 246 (25), 149 (16), 132 (3), 70 (100).




{kind=link}
{kind=link}