Introduction
Echinacea angustifolia,
Echinacea pallida and
Echinacea purpurea are the main medicinal
Echinacea species and have long been used to treat infections, to aid in wound healing and to enhance the immune system [
1]. In 2005,
Echinacea products ranked among the top botanical supplements sold in the United States. In recent years, treatment of rhinoviruses has been the focus of several studies, a number of which have failed to show the efficacy of
Echinacea [
2]. Commercial
Echinacea products often are mixtures of the three main medicinal species and there is no regulation of the concentrations of the chemical constituents. Among the chemical constituents of
Echinacea species, the alkamides, caffeic acid derivatives such as chicoric acid and the polyphenols are considered important for biological activity [
3].
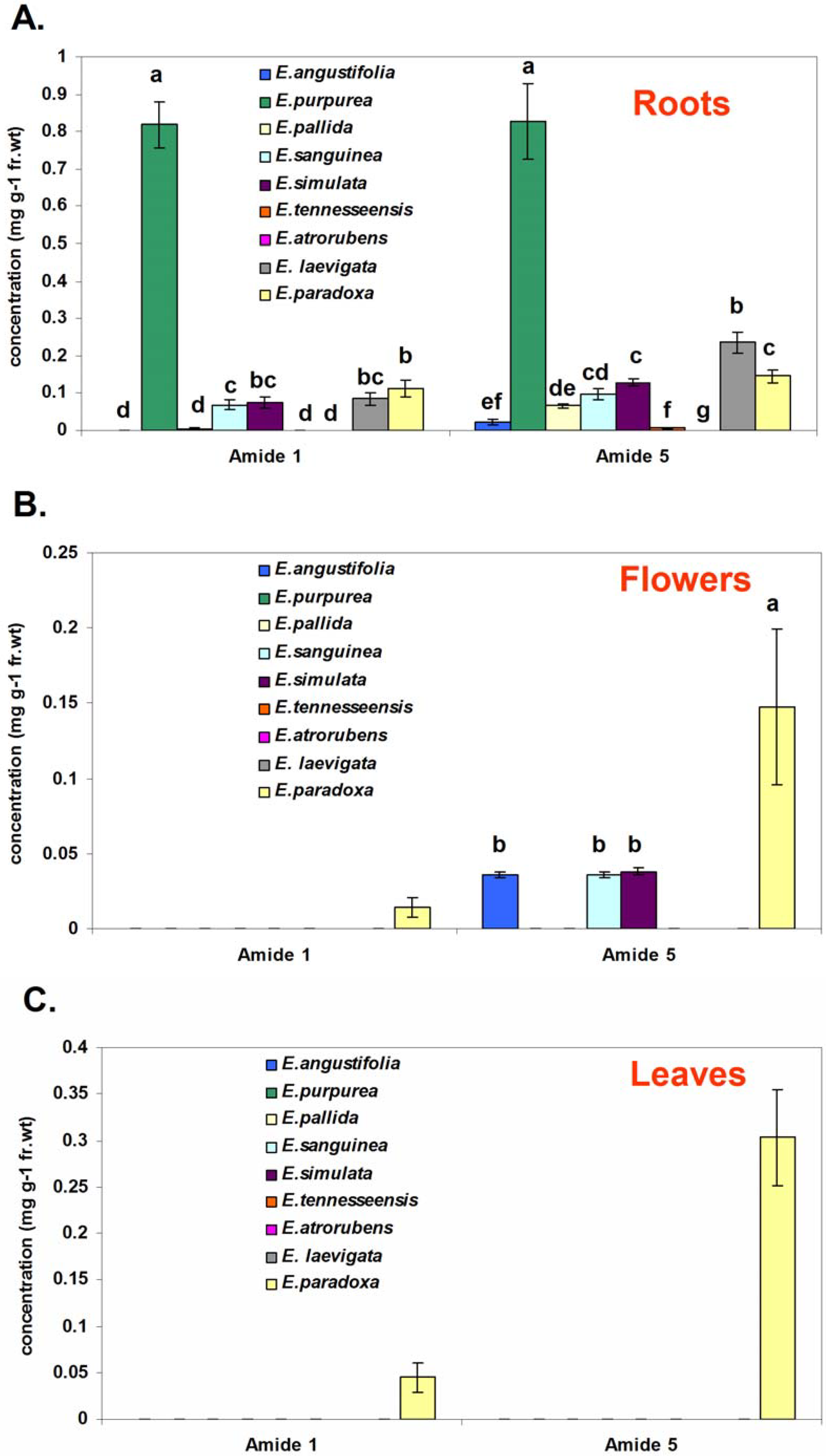
Alkamide levels differ significantly among roots, stems, and flowers of
E. purpurea. The roots had higher levels of the dodeca-2,4-diene-8,10-diyne alkamides, whereas levels of the dodecatetraene alkamides and nonadeca-2,4-diene-8,10-diynes were highest in stems [
4]. Accessions from different geographical regions often show different chemical fingerprints. Additionally, preliminary studies on the stability of alkamide compounds in
E. angustifolia revealed a 13% loss of alkamide levels over two months [
5]. The effects of storage time and temperature on alkamide levels in
E. purpurea roots showed that levels of all alkamides fell by over 80% during storage at 24° C for 64 weeks [
6]. The ready availability of authentic standards of select alkamides would not only facilitate standardization for the purposes of medical studies, but would also permit biological evaluation of individual components.
The recent discovery that dodecadiendiynoic amide
1 from
E. purpurea and
E. pallida inhibited LPS-mediated activation of a murine macrophage line, RAW264.7, suggests that this alkamide may have anti-inflammatory activity [
7].
Using male rats, an
in vivo study examined the immunomodulatory effects of alkamides purified from
Echinacea purpurea. These results suggest that the alkamides are among the active constituents of
E. purpurea plants. At a dose level of approximately 12 μg/kg body weight/day they effectively stimulated alveolar macrophage function in healthy rats [
8]. Alkamides isolated from
Echinacea angustifolia had inhibitory activity in
in vitro cyclooxygenase (sheep microsomes) and 5-lipoxygenase (porcine leukocytes) assays [
9]. Hexane extracts of
Echinacea variably inhibit growth of yeast strains of
Saccharomyces cerevisiae,
Candida shehata,
C. albicans, and
C. tropicalis under near UV irradiation and to a lesser extent without irradiation [
10]. Synergistic antioxidant effects were found when cichoric acid was combined with a natural mixture of alkamides [
11].
Experimental
General
Unless stated otherwise, all reactions were magnetically stirred and monitored by thin-layer chromatography (TLC) using 0.25 mm precoated silica gel F254 plates (Sigma-Aldrich). Column or flash chromatography were performed with the indicated solvents using silica gel (230-400 mesh) purchased from Dynamic Adsorbents, LLC. All melting points were obtained on a Laboratory Devices capillary melting point apparatus and are uncorrected. 1H- and 13C-NMR spectra were recorded on a Bruker VXR-300 (300 MHz) or a Bruker VXR-400 (400 MHz) spectrometer. Chemical shifts are reported relative to internal chloroform (1H, 7.26 ppm; 13C, 77.23 ppm). High resolution mass spectra were performed at the Iowa State University Mass Spectrometry Laboratory.
Plant material and extraction
Nine species of
Echinacea,
E. angustifolia (Accession 631267),
E. purpurea (Accession 631307),
E. pallida (Accession 631293),
E. sanguinea (Accession A23878),
E. simulata (Accession 631249),
E. tennesseensis (Accession 631325),
E. atrorubens (Accession 631262),
E. laevigata (Accession 631312)
and E. paradoxa (Accession 631301), provided by Dr. Mark P. Widrlechner at the USDA-ARS North Central Regional Plant Introduction Station, were studied to evaluate the natural distribution of amides
1 and
5 in
Echinacea species. Six-month-old roots, flowers and leaves from each species/accessions were used. Specific plant growth conditions, plant material harvest and extraction method are the same as those in our previously published work [
15]. 7-Hydroxy-(
E)-
N-isobutylundeca-2-ene-8,10-diynamide (C
15H
21O
2) was added as an internal standard prior to extraction for quantification purposes. All experiments were performed in triplicate on independently extracted plant samples from three individual plants.
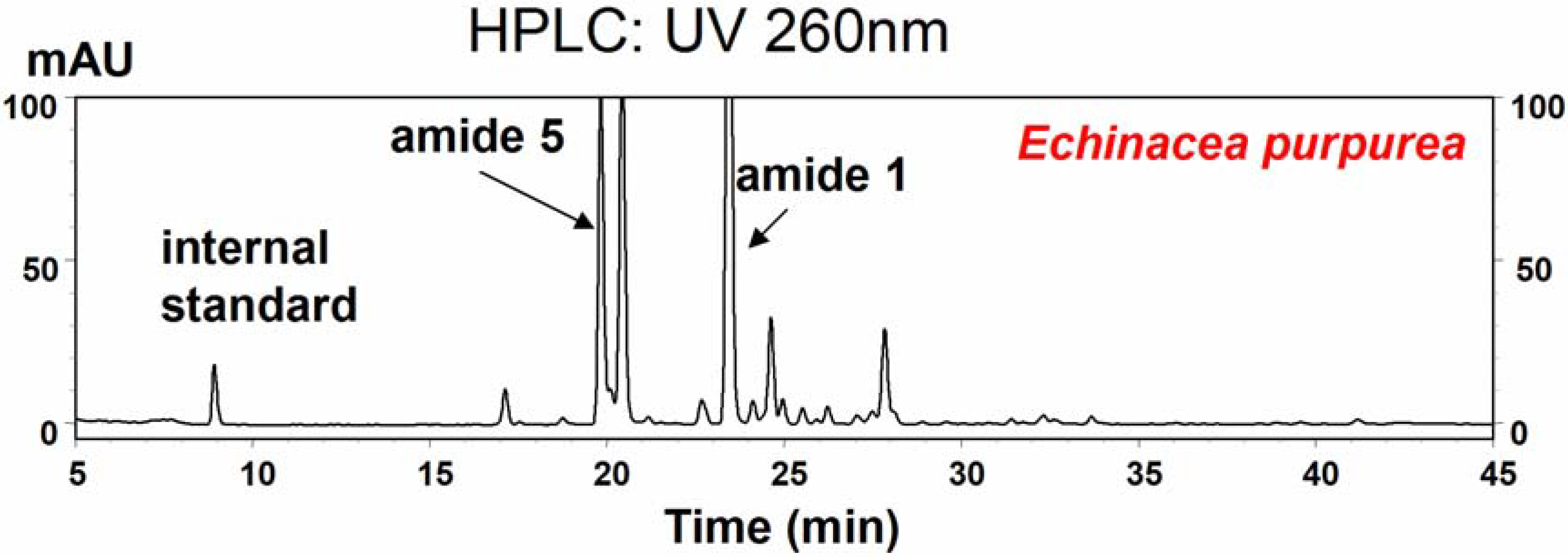
HPLC analysis
Ethanol extract (15 μL) was injected into a YMC-Pack ODS-AM RP C18 (250 x 4.6 mm, 5 μm) column (Waters, MA) on a Beckman Coulter HPLC equipped with a 508 autosampler, 126 pump control and 168 UV-photodiode array detector (PDA) controlled by 32karat TM software (Version 5.0). The solvent system used was CH3CN/H2O at a flow rate of 1.0 mL/min following a linear gradient of 40→80% CH3CN in H2O over 45 min. Online UV spectra were collected between 200–400 nm. Compound quantification was carried out by calculating the UV response relative to the internal standard 7-hydroxy-(E)-N-isobutylundeca-2-ene-8,10-diynamide (C15H21O2), which has been found suitable for use as an internal standard for these two amides because it was not found in Echinacea plants and does not overlap with any other metabolites found in Echinacea. Amides 1 and 5 were quantified at UV 260 nm with respect to the internal standard, using relative response factors to correct for absorbance differences between these two amides and the standard. These relative response factors for amides 1 and 5 were calculated at UV 260 nm. Various amount of authentic amide 1 or amide 5 (0.625 – 3.125 µg) with internal standard (2.5 µg) were injected to give average relative response factors of 0.0677 (R2 = 0.99) for amide 1, and 0.0669 (R2 = 0.99) for amide 5, respectively. The internal quantification method used here can account for variations in extraction efficiencies in different extracts. The HPLC detection limit for both of the amides was approximately 0.02 μg mL−1.
Statistical analysis
Statistical analyses were performed using SAS software version 8.02 (SAS Institute Inc., Cary, NC). One-way analysis of variance followed by the Tukey test was used to compare means. Significance of difference was defined at p < 0.05.
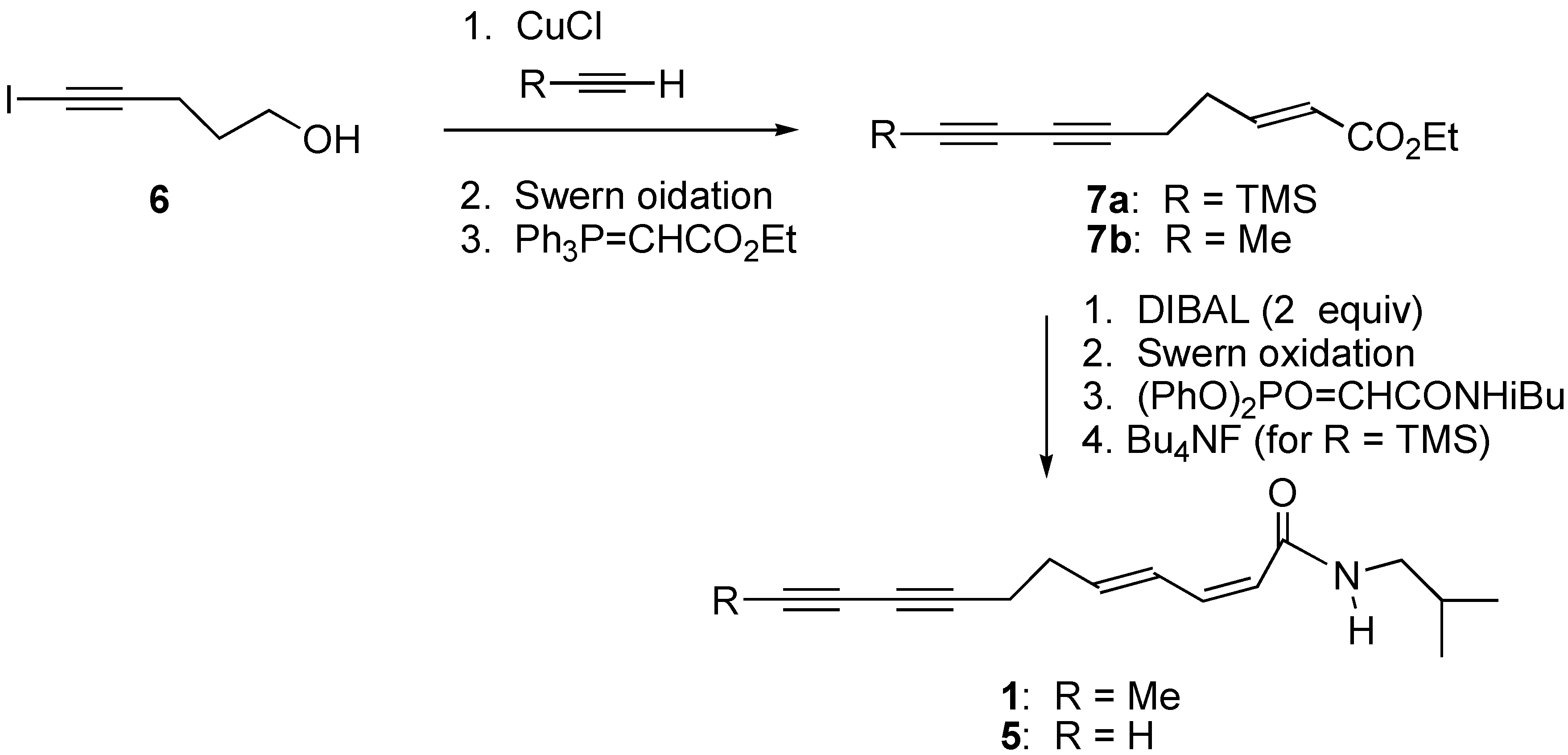
Ethyl 9-trimethylsilylnona-2-ene-6,8-diynoate (7a).
To a solution of trimethylsilylacetylene (0.5 mL, 3.51 mmol) and 5-iodo-4-pentynol 6 (0.281 g, 1.34 mmol) in degassed piperidine (2 mL) was added CuCl (0.014 g, 0.14 mmol) at 0 oC. The mixture was stirred at rt for 0.5 h and then quenched with sat. NH4Cl (aq) (6 mL) and extracted with Et2O (3 x 10 mL). The combined organic layers were washed with brine (2 x 20 mL), dried (MgSO4), filtered and concentrated. The crude residue was purified via flash chromatography to give the alcohol (0.188 g, 78 % yield).
Dimethylsulfoxide (0.766 mL, 10.8 mmol) was added dropwise at -78 oC to a solution of oxalyl chloride (0.471 mL, 5.4 mmol) in CH2Cl2 (10 mL). The mixture was stirred at same temp. for 20 min and triethylamine (2.25 mL, 16.2 mmol) was added dropwise and stirred at same temp. for 20 min. To the mixture was added the alcohol synthesized above (0.487 mg, 2.7 mmol) at -78 oC and stirred for 80 min while slowly warmed to room temperature. The reaction was quenched with sat NH4Cl (aq) and the aqueous layer was extracted with CH2Cl2 (2 x 20 mL). The combined organic layers were washed with water (2 x 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the aldehyde (0.409 g, 85 % yield).
To a solution of carbethoxymethyl(triphenyl)phosphonium bromide (3.94 g, 9.19 mmol) in THF (30 mL) was added n-BuLi (3.67 mL, 2.5 M soln in hexane) at 0 oC under Ar. The mixture was stirred for 20 min at 0 oC and added the above aldehyde (0.409 g, 2.29 mmol) at the same temperature. After 1 h of stirring at room temperature, reaction was quenched with sat. NH4Cl (aq) and extracted with Et2O (3x30 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give compound 7a (0.465 g, 82% yield). 1H-NMR (300 MHz, CDCl3) δ 6.94-6.89(m, 1H), 5.86 (d, J= 15.2 Hz, 1H), 4.17 (q, J= 7.2 Hz, 2H), 2.43 (m, 4H), 1.28 (t, J= 7.2 Hz, 3H), 0.18 (s, 9H).
2Z,4E-undeca-2,4-dien-8,10-diynoic acid isobutyl amide (5).
To a solution of compound 7a (0.341 g, 1.37 mmol) in 10 mL of THF was added DIBAL (4.12 mL, 1 M soln) at -78 oC in Ar. After stirring for 2 h at -78 oC, the reaction was quenched with EtOAc (30 mL) at -78 oC and the reaction wixture was warmed to rt, washed with 10% HCl(aq), brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash chromatography to give allyl alcohol (0.260 g, 92 % yield)
Dimethylsulfoxide (0.178 mL, 2.46 mmol) was added dropwise at -78 oC to a solution of oxalyl chloride (0.110 mL, 1.23 mmol) in CH2Cl2 (5 mL). The mixture was stirred at the same temperature for 20 min and triethylamine (0.526 mL, 3.69 mmol) was added dropwise and stirred at same temperature for 20 min. To the mixture was added the above alcohol (0.127 mg, 0.616 mmol) at -78 oC and stirred for 80 min while slowly warming to rt. The reaction was quenched with sat NH4Cl (aq) and aqueous layer was extracted with CH2Cl2 (2x 10 mL). The combined organic layers were washed with water (2x 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give aldehyde (0.106 g, 81% yield.)
To a solution of diphenylphosphonoacetamide (0.187 g, 0.539 mmol) in 10 mL of THF was added NaHMDS (0.735 mL, 1M soln in THF) at -78 oC and stirred at same temperature for 20 min. To the mixture was added above aldehyde (0.1 g, 0.49 mmol) in THF (2 mL) by cannula and the resulting mixture was warmed to 10 oC over 2 h. The reaction was quenched with NH4Cl (aq), washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give (2Z, 4E) amide (0.090 g, 62 % yield). 1H-NMR (300 MHz, CDCl3) δ 7.49 (dd, J= 15.3, 11.4 Hz, 1H), 6.37 (t, J= 11.4 Hz, 1H), 6.05-5.90 (m, 1H), 5.58 (brs, 1H), 5.52 (d, J= 12.9 Hz, 1H), 3.12 (t, J= 6.9 Hz, 2H), 2.39-2.38 (m, 4H), 1.84-1.75 (m, 1H), 0.92 (d, J= 6.9 Hz, 6H), 0.18 (s, 9H); 13C-NMR (75 MHz, CDCl3) δ 166.5, 140.9, 140.0, 128.5, 119.9, 88.5, 82.3, 79.1, 66.2, 46.9, 31.6, 28.8, 20.4, 19.4, -0.13; HRMS m/e (EI) for C18H27NOSi (M)+ calcd 301.1862, measured 301.1843
To a solution of the above (2Z, 4E) amide (0.032 g, 0.106 mmol) in THF (2 mL) was added TBAF (0.159 mL, 1.159 mmol) at 0 oC. The mixture was stirred for 1h at rt and the solvent was removed. The crude residue was purified via flash column chromatography to give compound 5 (0.024 g, 99 % yield) 1H-NMR (300 MHz, CDCl3) δ 7.51 (dd, J= 14.7, 11.4 Hz, 1H), 6.37(t, J= 11.4 Hz, 1H), 6.02-5.89 (m, 1H), 5.63 (brs, 1H), 5.53 (d, J= 11.4 Hz, 1H), 3.12 (t, J= 6.6 Hz, 2H), 2.49-2.31 (m, 4H), 1.97 (s, 1H), 1.84-1.74 (m, 1H), 0.91 (d, J= 6.6 Hz, 6H);13C-NMR (75 MHz, CDCl3) δ 166.5, 140.9, 139.8, 128.5, 119.9, 82.3, 77.5, 65.2, 65.1, 46.9, 31.4, 28.8, 20.4, 19.1; HRMS m/e (EI) for C15H19NO (M)+ calcd 229.1467, measured 229.1579.
Ethyl deca-2-ene-6,8-diynoate (7b).
Degassed piperidine (5.5 mL), 5-iodo-4-pentynol (1.74 g, 8.49 mmol) and CuCl (0.086 g, 0.85 mmol) were mixed in a sealed tube. The mixture was cooled to -78 oC and excess propyne gas (condensed to liquid, 2 mL) was added by blowing along the wall of the tube. The mixture was slowly warmed to room temperature. After stirring for 2 h at rt, the mixture was cooled to -78 oC and the sealed tube was opened then slowly warmed to rt to evaporate excess propyne. NH4Cl (aq) (20 mL) was added to the mixture then extracted with Et2O (3 x 20 mL). The organic layer was washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give alcohol (0.847 g, 82 % yield)
Dimethylsulfoxide (1.63 mL, 22.9 mmol) was added dropwise at -78 oC to a solution of oxalyl chloride (1 mL, 11.5 mmol) in 60 mL of CH2Cl2. The mixture was stirred at the same temperature for 20 min and triethylamine (4.78mL, 34.4mmol) was added dropwise and stirred at same temperature for 20 min. To the mixture was added above alcohol (0.70 g, 5.73 mmol) at -78 oC and stirred for 80 min while slowly warmed to rt. The reaction was quenched with sat NH4Cl (aq) (10 mL) and aqueous layer was extracted with CH2Cl2 (2x 30 mL). The combined organic layers were washed with water (2x 20 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give aldehyde (0.55 g, 80 % yield). 1H-NMR (300 MHz, CDCl3) δ 9.76 (s, 1H), 2.68 (t, J= 6.6 Hz, 2H), 2.54 (t, J=6.6 Hz, 2H), 1.89 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 199.9, 74.4, 74.2, 66.5, 64.4, 42.4, 12.6, 4.3.
To a solution of carbethoxymethyl(triphenyl)phosphonium bromide (5.26 g, 12.37 mmol) in THF (40 mL) was added n-BuLi (4.95 mL, 2.5 M soln in hexane) at 0 oC under Ar. The mixture was stirred for 20 min at 0 oC and the aldehyde (0.59 g, 4.95 mmol) was added at same temperature. After 1 h of stirring at rt, the reaction was quenched with sat NH4Cl (aq) and extracted with ethyl ether (3x30 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give compound 7b (0.73 g, 78 % yield). 1H-NMR (300 MHz, CDCl3) δ 7.01-6.85 (m, 1H), 5.86 (d, J= 15.6Hz, 1H), 4.18 (q, J= 7.2 Hz, 2H), 2.43-2.40 (m, 4H), 1.90 (s, 3H), 1.26 (t, J= 7.2 Hz, 3H).
2Z,4E-undeca-2,4-dien-8,10-diynoic acid isobutyl amide (1).
To a solution of compound 7b (0.437 g, 2.3 mmol) in 20 mL of THF was added DIBAL (4.6 mL,1.0M soln in THF) at -78 oC in Ar. After stirring for 2 h at -78oC, the reaction was quenched with ethyl acetate (30 mL) at -78 oC and warmed to rt. The mixture was washed with 10% HCl (aq) (10 mL), brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give the allylic alcohol (0.28 g, 81 % yield).
Dimethylsulfoxide (0.530 mL, 7.48 mmol) was added dropwise at -78 oC to a solution of oxalyl chloride (0.326 mL, 3.74 mmol) in 20 mL of CH2Cl2. The mixture was stirred at the same temp for 20 min and triethylamine (1.56 mL, 11.2 mmol) was added dropwise and stirred at same temperature for 20 min. The above alcohol (0.277 g, 1.87 mmol) was added to the mixture at -78 oC and stirred for 80 min while slowly warming to rt. The reaction was quenched with sat NH4Cl (aq) and aqueous layer was extracted with CH2Cl2 (2x 10 mL). Combined organic layer was washed with water (2x 10 mL), dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give aldehyde (0.229 g, 84 % yield). 1H-NMR (300 MHz, CDCl3) δ 9.49 (d, J= 7.8 Hz, 1H), 6.83 (dt, J= 15.6, 6.0 Hz, 1H), 6.14 (dd, J= 15.6, 7.8 Hz, 1H), 2.58-2.40 (m, 4H), 1.86 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 193.9, 155.4, 134.0, 74.3, 74.3, 67.0, 60.6, 31.4, 18.2, 4.3.
To a solution of diphenylphosphonoacetamide (0.370 g, 1.06 mmol) in THF (10 mL) was added NaHMDS (1.06 mL, 1 M soln in THF) at -78 oC and stirred at same temp for 20 min. To the mixture was added the above aldehyde (0.140 g, 0.97 mmol) in THF (2 mL) via cannula and the resulting mixture was warmed to 10 oC over 2h. The reaction was quenched with NH4Cl (aq), washed with water, brine, dried (MgSO4), filtered and concentrated in vacuo. The crude residue was purified via flash column chromatography to give amide 1 (0.131g, 56% yield). 1H-NMR (300 MHz, CDCl3) δ 7.47 (dd, J= 15.3, 11.4 Hz, 1H), 6.34 (t, J= 11.4 Hz, 1H), 5.99-5.87 (m, 1H), 5.78 (brs, 1H), 5.52 (d, J= 11.4 Hz, 1H), 3.09 (t, J= 6.6 Hz, 2H), 2.37-2.32 (m, 4H), 1.87 (s, 3H), 1.82-1.73 (m, 1H), 0.89 (d, J= 6.6 Hz, 6H); 13C-NMR (75 MHz, CDCl3) δ 166.7,140.9, 140.3, 128.4, 119.9, 75,8, 73,7, 66.2, 64.7, 46.9, 31.8, 28.8, 20.4, 19.3, 4.4.

{kind=link}
{kind=link}
{kind=link}




