Synthesis, Characterization, and Saccharide Binding Studies of Bile Acid − Porphyrin Conjugates

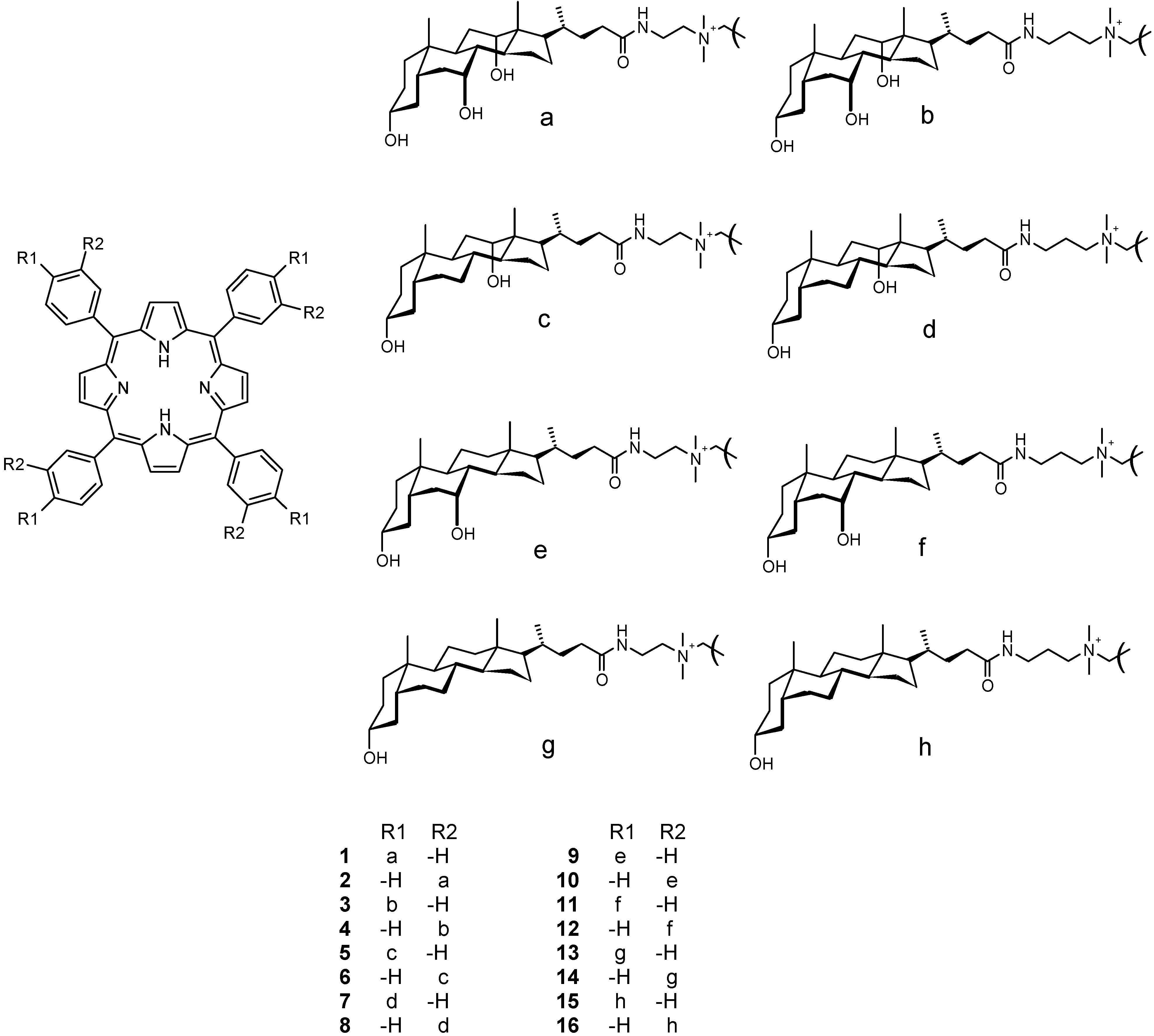{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction

Results and Discussion
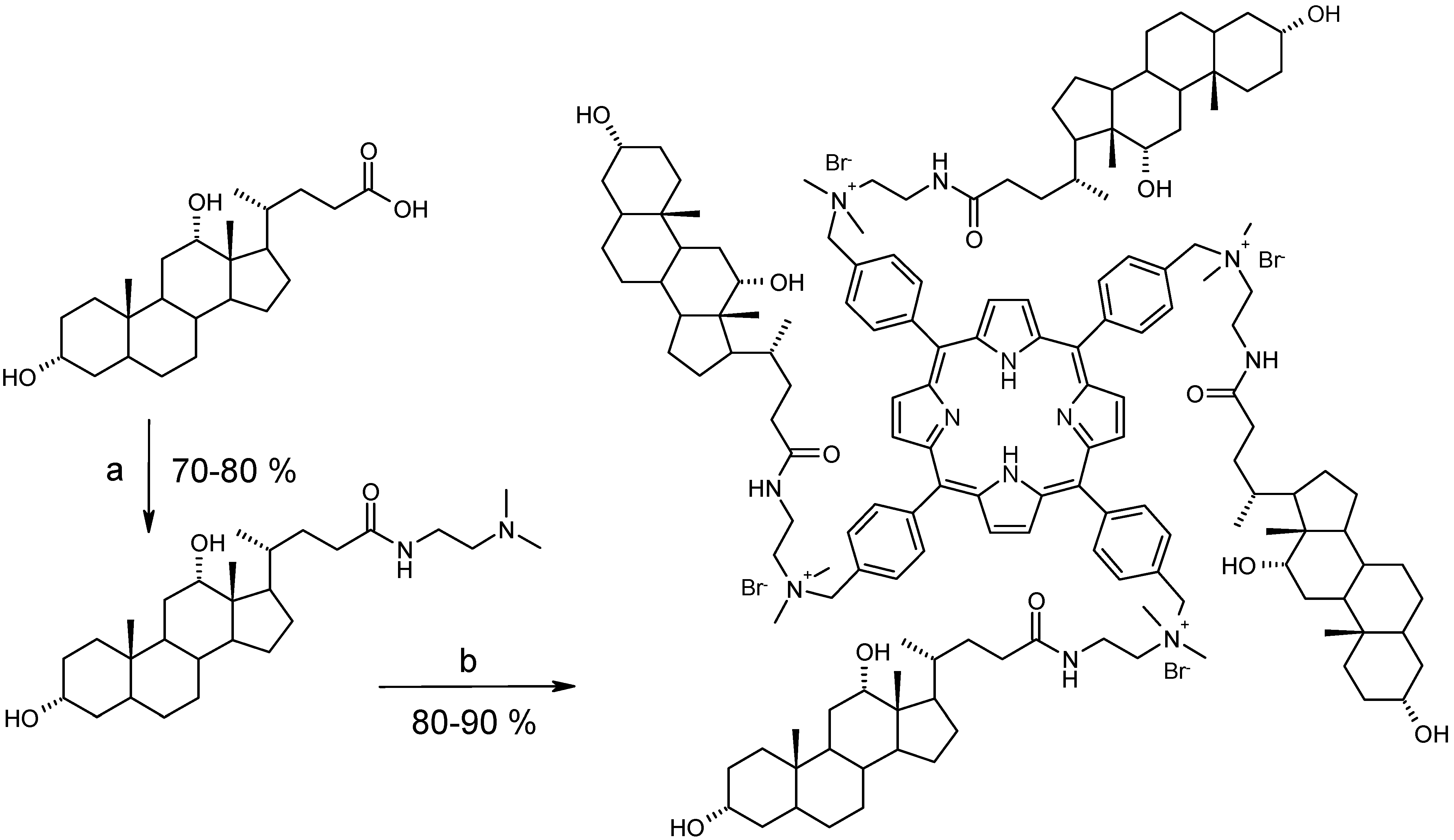
Synthesis and characterization


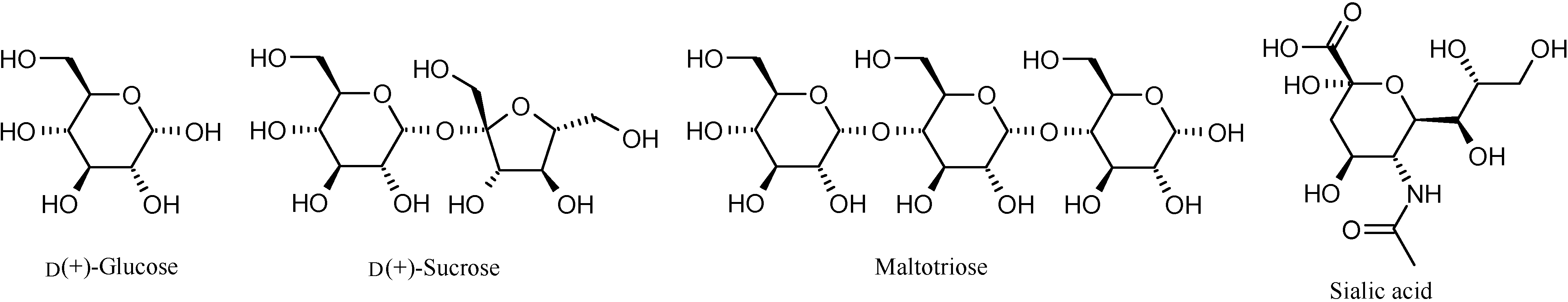
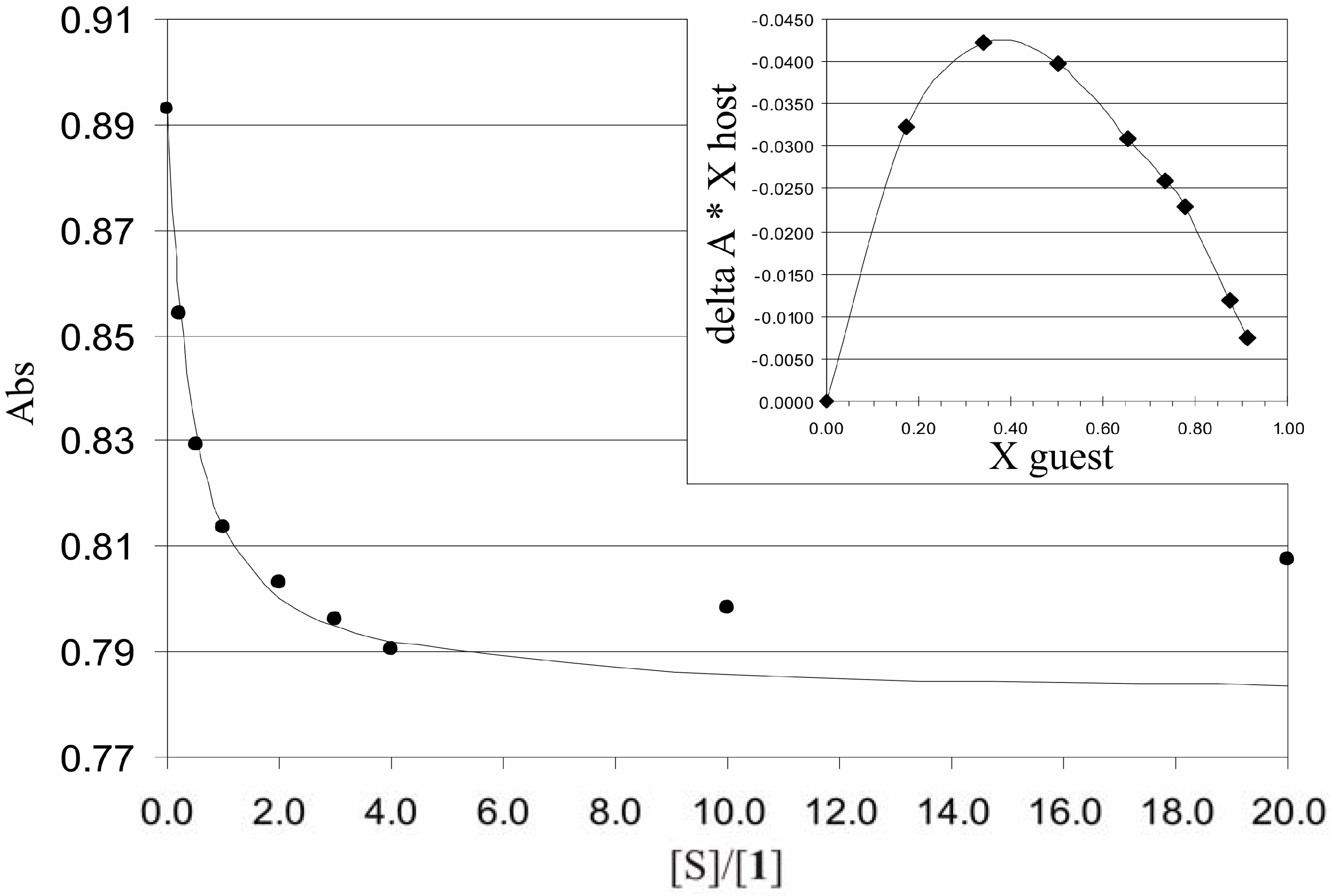
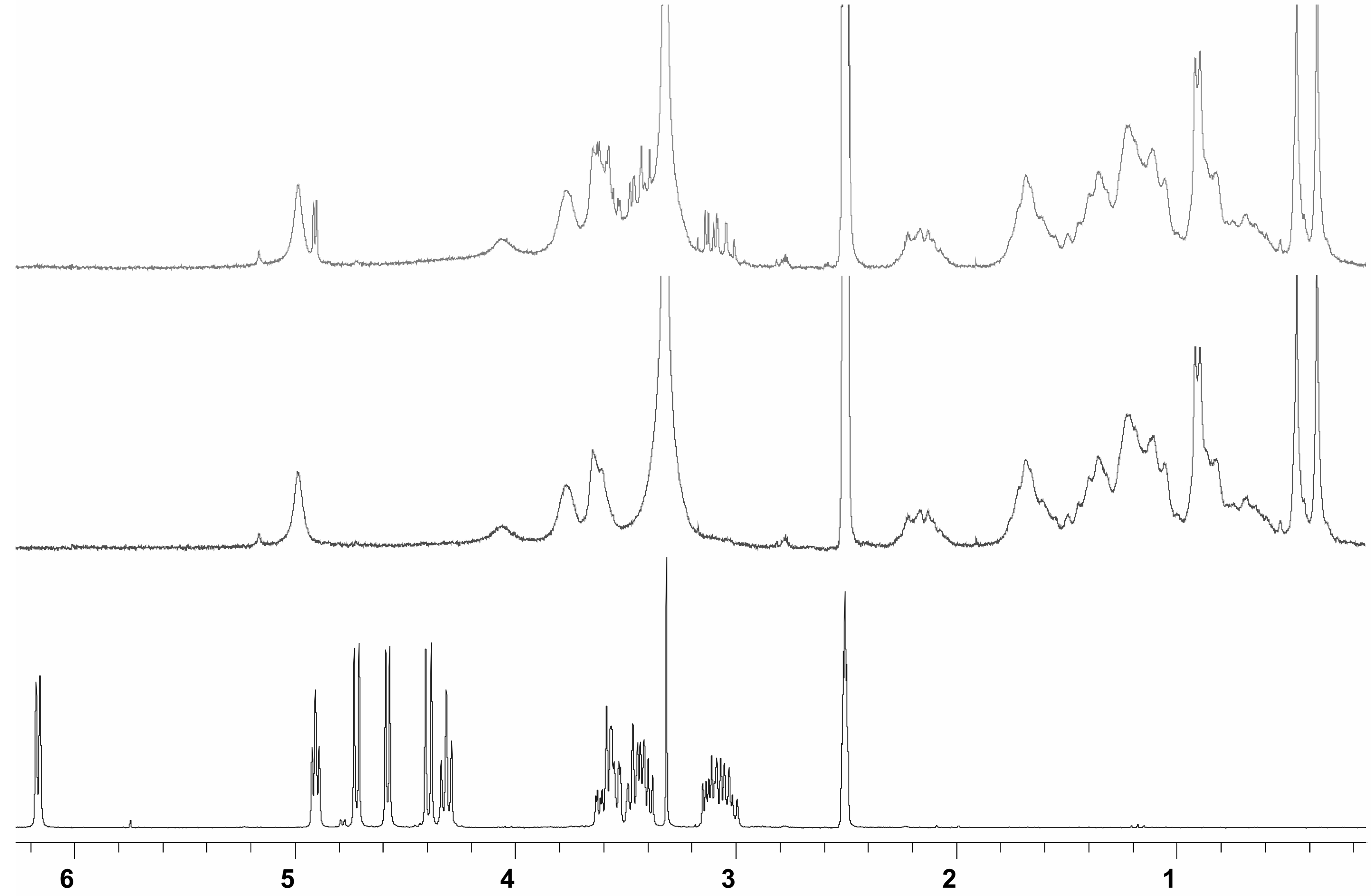
Binding studies



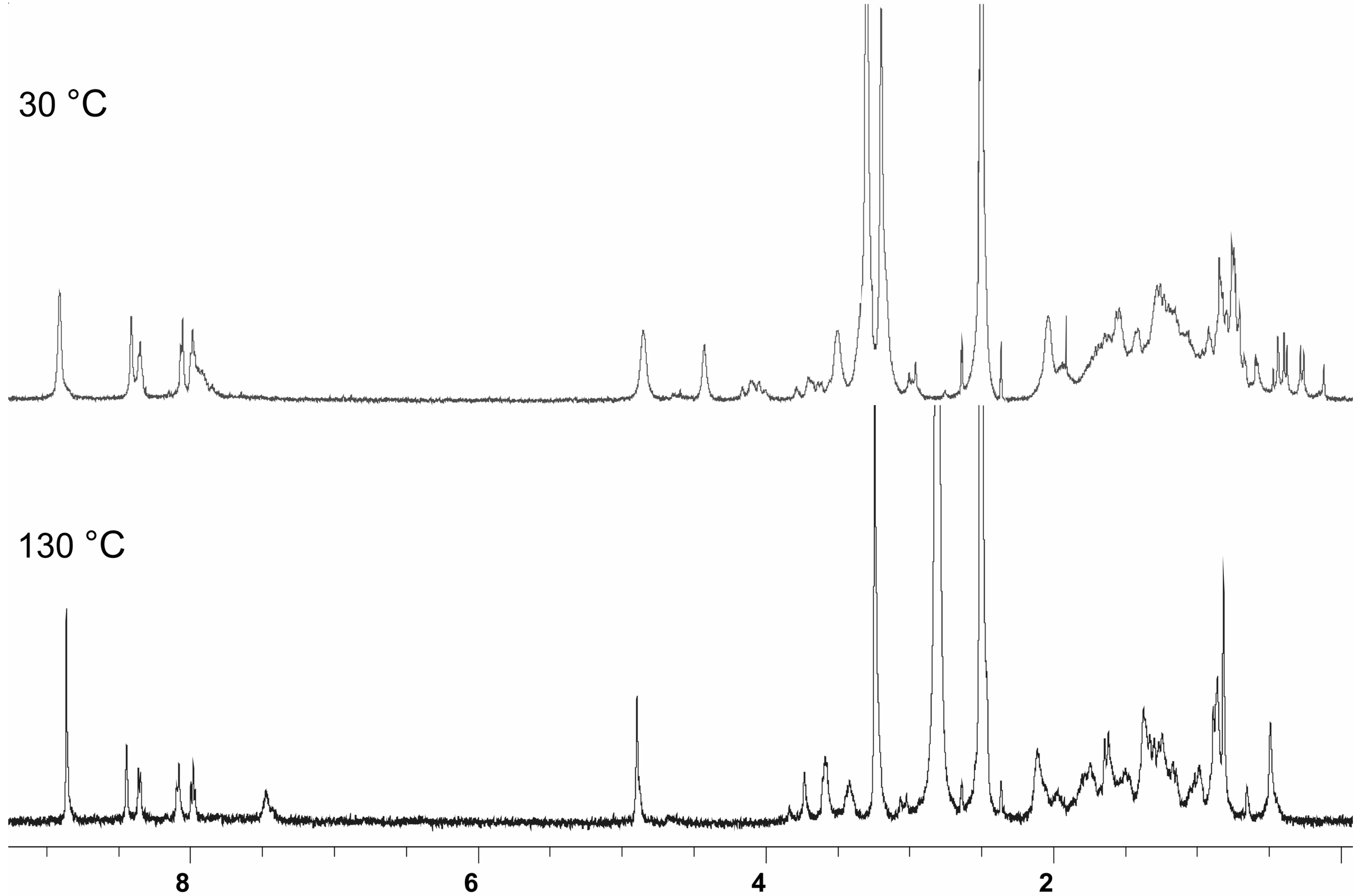
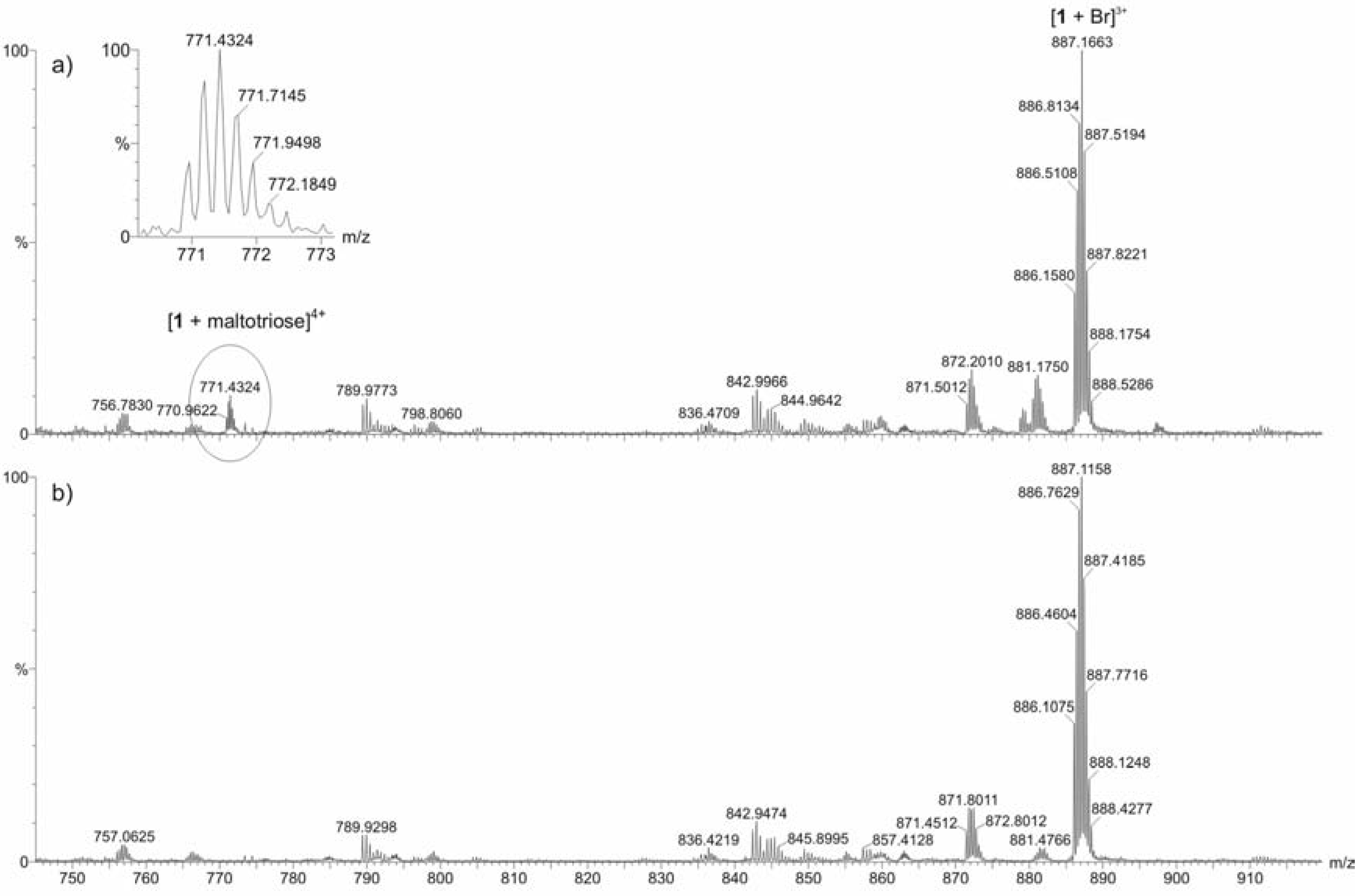
ESI mass spectrometric measurements


Conclusions
Experimental
General
Analytical and spectral data for 5-16.
Acknowledgements
References and Notes
- Virtanen, E.; Kolehmainen, E. Use of bile acids in pharmacological and supramolecular applications. Eur. J. Org. Chem. 2004, 3385–3399, and references cited therein. [Google Scholar]
- Enhsen, A.; Kramer, W.; Wess, G. Bile acids in drug discovery. Drug Discov. Today 1998, 3, 409–418. [Google Scholar] [CrossRef]
- Hofmann, A.F. Bile acids as drugs: principles, mechanisms of action and formulation. Ital. J. Gastroenterol. 1995, 27, 106–113. [Google Scholar]
- Matile, S.; Som, A.; Sordé, N. Recent synthetic ion channels and pores. Tetrahedron 2004, 60, 6405–6435. [Google Scholar] [CrossRef]
- Janout, V.; Jing, B.; Regen, S.L. Molecular Umbrella-Assisted Transport of an Oligonucleotide across Cholesterol-Rich Phospholipid Bilayers. J. Am. Chem. Soc. 2006, 127, 15862–15870. [Google Scholar] [CrossRef]
- Dukh, M.; Šaman, D.; Lang, K.; Pouzar, V.; Černý, I.; Drašar, P.; Král, V. Steroid–porphyrin conjugate for saccharide sensing in protic media. Org. Biomol. Chem. 2003, 1, 3458–3463. [Google Scholar] Charvatová, J.; Rusin, O.; Král, V.; Volka, K.; Matejka, P. Novel porphyrin based receptors for saccharide recognition in water. Sens. Actuators, B: Chem. 2001, 76, 366–372. [Google Scholar] [CrossRef]
- Hirata, O.; Kubo, Y.; Takeuchi, M.; Shinkai, S. Mono- and oligosaccharide sensing by phenylboronic acid-appended 5,15-bis(diarylethynyl)porphyrin complexes. Tetrahedron 2004, 60, 11211–11218. [Google Scholar] [CrossRef] Hirata, O.; Yamamoto, M.; Sugiyasu, K.; Kubo, Y.; Ikeda, M.; Takeuchi, M.; Shinkai, S. Allosteric saccharide sensing by a phenylboronic-acids-appended 5,15-bis(triaryl-ethynyl)porphyrin. J. Supramol. Chem. 2003, 2, 133–142. [Google Scholar]
- Mizutani, T.; Kurahashi, T.; Murakami, T.; Matsumi, N.; Ogoshi, H. Molecular recognition of carbohydrates by zinc porphyrins: Lewis acid/Lewis base combinations as a dominant factor for their selectivity. J. Am. Chem. Soc. 1997, 119, 8991–9001. [Google Scholar] [CrossRef]
- Nakata, E.; Nagase, T.; Shinkai, S.; Hamachi, I. Coupling a natural receptor protein with an artificial receptor to afford a semisynthetic fluorescent biosensor. J. Am. Chem. Soc. 2004, 126, 490–495. [Google Scholar] [CrossRef]
- Kim, Y-H.; Hong, J-I. Carbamate-appended Zn-porphyrin: a neutral receptor for anions. Tetrahedron Lett. 2000, 41, 4419–4423. [Google Scholar] [CrossRef]
- Lee, C.; Lee, D.H.; Hong, J-I. Colorimetric anion sensing by porphyrin-based anion receptors. Tetrahderon Lett. 2001, 42, 8665–8668. [Google Scholar] [CrossRef]
- Starnes, S.D.; Arungundram, S.; Saunders, C.H. Anion sensors based on β,β’-disubstituted porphyrin derivatives. Tetrahedron Lett. 2002, 43, 7785–7788. [Google Scholar] [CrossRef]
- Kejík, Z.; Záruba, K.; Michalík, D.; Šebek, J.; Dian, J.; Pataridis, S.; Volka, K.; Král, V. Optical sensing system for ATP using porphyrin–alkaloid conjugates. Chem. Commun. 2006, 1533–1535. [Google Scholar]
- Král, V.; Shishkanová, T.V.; Sessler, J.L.; Brown, C.T. Cytosine-substituted metalloporphyrins: receptors for recognition of nucleotides in ion-selective electrodes. Org. Biomol. Chem. 2004, 2, 1169–1175. [Google Scholar] [CrossRef]
- Kolehmainen, E.; Koivukorpi, J.; Sievänen, E.; Král, V. Novel porphyrin-cholic acid conjugates as receptors for biologically important anions. Supramol. Chem. 2005, 17, 437–441. [Google Scholar] [CrossRef]
- Král, V.; Vasek, P.; Cigler, P.; Králová, J.; Poucková, P. Preparation of porphyrin derivatives with polymethine substitution for photodynamic therapy. Czech Patent 20022272, 2004. [Google Scholar]
- Kubát, P.; Lang, K.; Král, V.; Anzenbacher, P., Jr. Preprogramming of porphyrin-nucleic acid assemblies via variation of the alkyl/aryl substituents of phosphonium tetratolylporphyrins. J. Phys. Chem. B. 2002, 106, 6784–6792. [Google Scholar] [CrossRef] [Green Version]
- Corrigendum for ref 15: Kolehmainen, E.; Koivukorpi, J.; Sievänen, E.; Král, V. Novel porphyrin-cholic acid conjugates as receptors for biologically important anions. Supramol. Chem. 2005, 17, 437–441, Supramol. Chem. in press.. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1-16 are available from authors.
© 2007 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Koivukorpi, J.; Sievänen, E.; Kolehmainen, E.; Král, V. Synthesis, Characterization, and Saccharide Binding Studies of Bile Acid − Porphyrin Conjugates. Molecules 2007, 12, 13-24. https://doi.org/10.3390/12010013
Koivukorpi J, Sievänen E, Kolehmainen E, Král V. Synthesis, Characterization, and Saccharide Binding Studies of Bile Acid − Porphyrin Conjugates. Molecules. 2007; 12(1):13-24. https://doi.org/10.3390/12010013
Chicago/Turabian StyleKoivukorpi, Juha, Elina Sievänen, Erkki Kolehmainen, and Vladimír Král. 2007. "Synthesis, Characterization, and Saccharide Binding Studies of Bile Acid − Porphyrin Conjugates" Molecules 12, no. 1: 13-24. https://doi.org/10.3390/12010013
APA StyleKoivukorpi, J., Sievänen, E., Kolehmainen, E., & Král, V. (2007). Synthesis, Characterization, and Saccharide Binding Studies of Bile Acid − Porphyrin Conjugates. Molecules, 12(1), 13-24. https://doi.org/10.3390/12010013