Results and Discussion
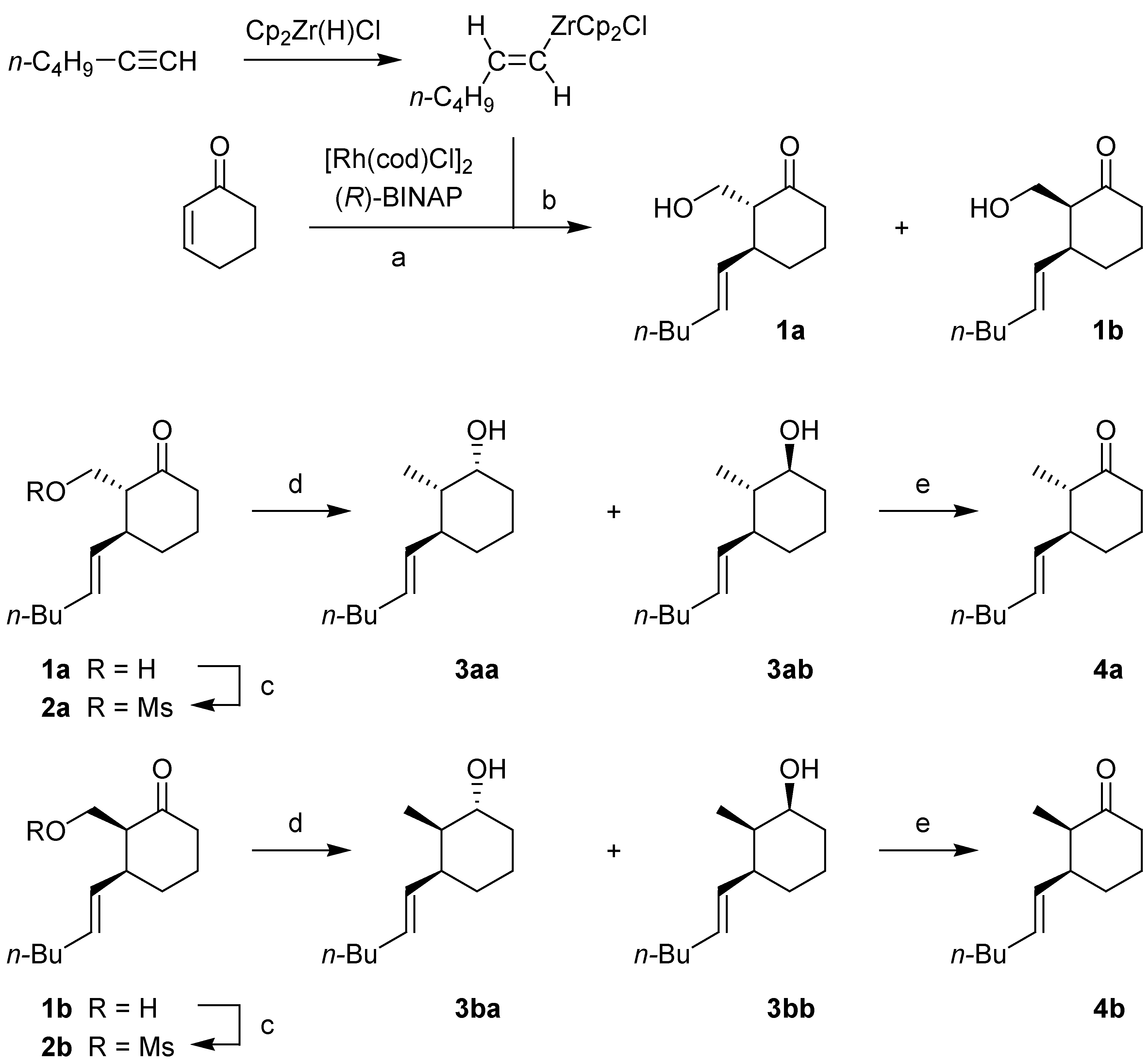
The synthesis of the two epimeric cyclohexanones 4a and 4b rests on the asymmetric 1,4-introduction of the hexenyl side chain on 2-cyclohexenone followed by trapping of the resulting metal O-enolate with an electrophilic reagent.
Scheme 1.
Synthetic pathway to epimeric (3S)-3-((E)-hex-1-enyl)-2-methylcyclo-hexanones 4a and 4b.
Scheme 1.
Synthetic pathway to epimeric (3S)-3-((E)-hex-1-enyl)-2-methylcyclo-hexanones 4a and 4b.
(a) Cp2Zr(H)Cl, n-C4H9C≡CH, THF, rt, 45 min; [Rh(cod)Cl]2, (R)-BINAP, THF, rt, 30 min; 2-cyclohexenone, rt, 3 h. (b) CH2O (from paraformaldehyde), –78 °C (89%). (c) MsCl, Et3N, CH2Cl2, 0 °C (a: 96%; b: 99%). (d) LiAlH4, Et2O, reflux, 2 h (a: 95%; b: 98%). (e) (COCl)2, DMSO, Et3N, –78 °C → rt (a: 89%; b: 96%).
Among several known enantioselective 1,4-additions of organometallic reagents to α,β-unsaturated ketones, we obtained the best results with the recently reported rhodium(I)-catalysed addition of alkenylzirconocene chlorides with BINAP as chiral ligand [
3,
4]. This result also follows Nicolaou’s report of the tandem reaction of the rhodium-catalysed asymmetric additions of alkenylzirconium reagents followed by trapping of the zirconium enolate by aldehydes [
5]. In our case, the 1,4-addition was performed on 2-cyclohexenone using (
E)-1-hexenylzirconocene chloride [
6], prepared from 1-hexyne and bis(cyclopentadienyl)zirconium chloride hydride (Cp
2Zr(H)Cl or Schwartz reagent) in the presence of a catalytic amount of the Rh(I)-complex [Rh(cod)Cl]
2 and (
R)-BINAP as a chiral ligand. As previously observed by Schwartz, we were unable to directly alkylate the intermediate zirconium
O-enolate [
7]; however, reaction with gaseous formaldehyde at –78 °C led, after acid work-up, to a 2.7:1 mixture (89% yield) of
1a and
1b, respectively [
8], with an excellent ee (better than 96%) [
9]. The obtained mixture was readily separated by flash chromatography. The assignment of the
trans- and
cis-relationship to the alkyl substituents in the
a- and
b-series, respectively, rests on the analysis of NMR spectral data of
1a. The diequatorial orientation in
1a follows from the large vicinal
J-value of 11.7 Hz for the coupling between H
a and H
b. The same coupling in epimer
1b (5.3 Hz) is indicative of a
cis-relationship (
Figure 1). The absolute configuration was assigned on the basis of the results obtained by Oi and Inoue [
3].
Figure 1.
1H-NMR structural assignment of 1a.
Figure 1.
1H-NMR structural assignment of 1a.
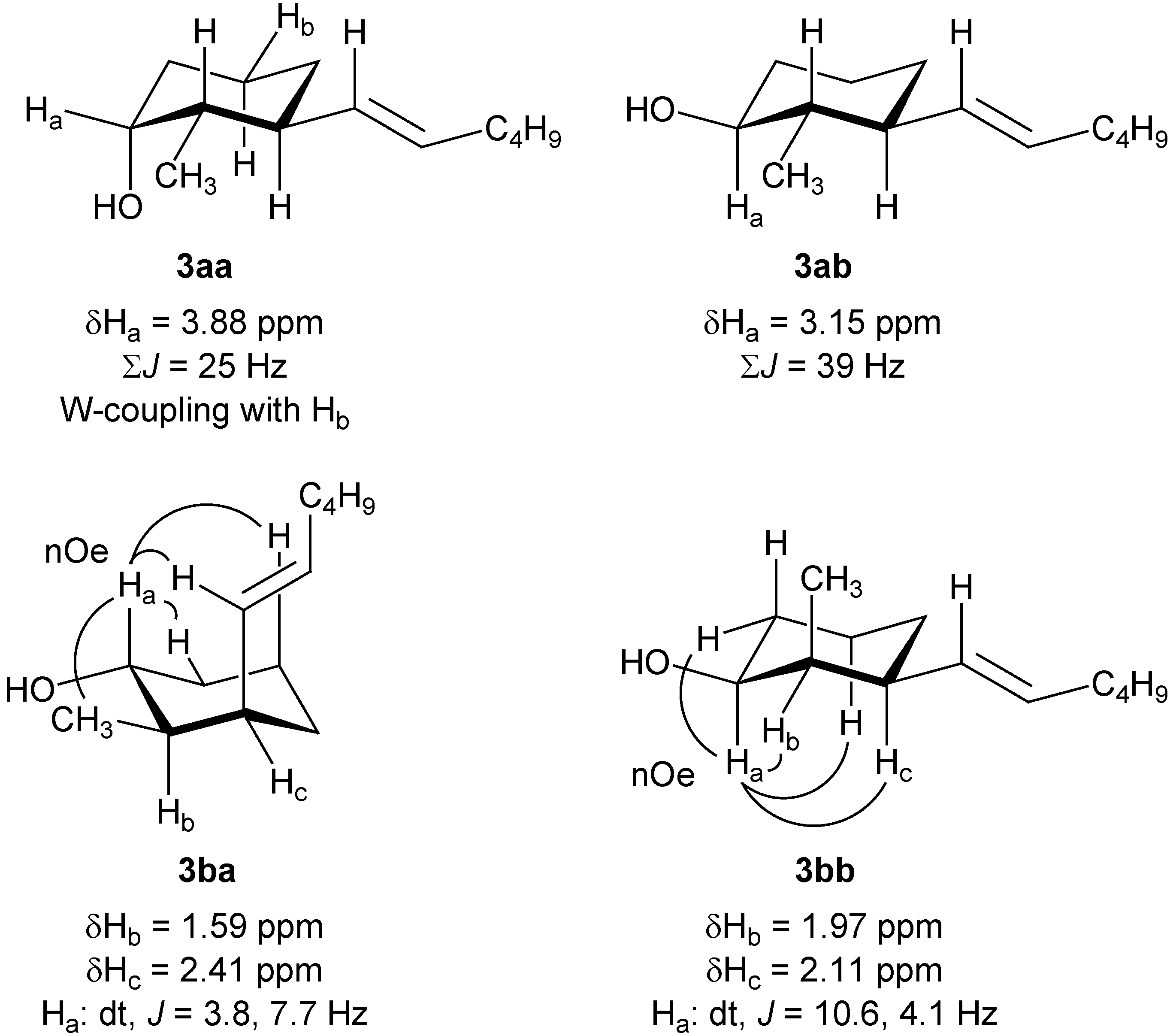
Figure 2.
1H-NMR structural assignments of the intermediate alcohols 3aa, 3ab, 3ba, and 3bb.
Figure 2.
1H-NMR structural assignments of the intermediate alcohols 3aa, 3ab, 3ba, and 3bb.
The further conversion of
1a and
1b to
4a and
4b, respectively, first involves the reduction of the mesylates
2a and
2b, obtained by treatment with mesyl chloride and triethylamine in dichloromethane, to afford a mixture of the two epimeric alcohols. From
2a there was obtained in 95% yield a 1:4 mixture of
3aa and
3ab; from
2b there was obtained in 98% yield a 1:4 mixture of
3ba and
3bb, respectively. Again the structural assignment of the four isomeric alcohols rested on
1H-NMR analysis (
Figure 2). Within the
trans-series distinction between
3aa and
3ab readily follows from the coupling constant pattern of the H
a proton, which indicates an axial hydroxyl group in
3aa (cf. smaller sum of vicinal
J-values for H
a) and an equatorial one in
3ab (cf. larger sum of vicinal
J-values for H
a). Furthermore a characteristic long range W coupling is observed between H
a and H
b in
3aa. The structural assignment in the
cis-series (
3ba and
3bb) on the other hand rests on the observed nOe’s in both derivatives, in particular between H
a and the olefinic proton of the hexenyl side-chain in
3ba. Finally, Swern oxidation of both mixtures led to the desired cyclohexanones
4a and
4b [
10].
Experimental
General
Dichloromethane was distilled from CaH2. Diethyl ether and tetrahydrofuran (THF) were distilled from benzophenone ketyl. TLC were run on glass plates precoated with silica gel (Merck, 60F-254). Column chromatography was performed on silica gel (Merck, 230-400 mesh). IR spectra (KBr films) were recorded on a Perkin–Elmer series 1600 FT-IR spectrometer. 1H-NMR and 13C-NMR spectra were recorded on a Bruker AM-500 spectrometer operating at 500 (1H) and 125 MHz (13C), respectively. Mass spectra (EI) were recorded on a Hewlett–Packard 5898A spectrometer at 70 eV.
(2R,3S)-3-((E)-Hex-1-enyl)-2-(hydroxymethyl)cyclohexanone (1a) and (2S,3S)-3-((E)-hex-1-enyl)-2-(hydroxylmethyl)cyclohexanone (1b).
Dry formaldehyde was prepared as follows: paraformaldehyde is predried overnight in vacuo at 60 °C in a three-necked 100-mL, round-bottom flask. The flask is equipped with an inlet for N2 (dried over molecular sieves) and is connected via Teflon tubing to the reaction flask. The latter is then equipped with a CaCl2 drying tube. The entire system is evacuated, filled with N2 and the dried paraformaldehyde is depolymerised in a stream of N2 by heating at 180 °C. To a suspension of Cp2ZrHCl (6.20 g, 24 mmol) in dry THF (80 mL) under Ar was added 1-hexyne (2.76 mL, 24 mmol) and the mixture was stirred at rt for 45 minutes to give a solution of 1-hexenyl-zirconocene chloride. In a two-necked 250-mL flask under Ar, [Rh(cod)Cl]2 (247 mg, 1 mmol) and (R)-BINAP (749 mg, 1.2 mmol) were dissolved in dry THF (40 mL) and the solution was stirred at rt for 0.5 h. To the solution of rhodium catalyst, cyclohexenone (1.94 mL, 20 mmol) and the solution of 1-hexenylzirconocene chloride were added and the mixture was stirred at rt for 3 h. The reaction mixture was cooled to –78 °C and quenched with gaseous dry formaldehyde (see above). After warming up the mixture to rt, an aqueous saturated NH4Cl solution (3 mL) was added and the resulting mixture was stirred for 0.5 h. t-Butylmethyl ether (MTBE; 200 mL) was added and the precipitate formed was removed by filtration. After removal of the solvent under reduced pressure, the residue was purified by flash chromatography on silica gel (n-pentane/EtOAc, 83:17 to 75:25) to give cyclohexanones 1a (2.70 g) and 1b (1.04 g) in 89% total yield.
1a: Rf (isooctane/EtOAc, 1:1) 0.38; [α]Drt –10.0 (c 1.0, CHCl3); IR ν 3493 (br, m), 2929 (vs), 2871 (vs), 1701 (vs), 1459 (m), 1328 (m), 1222 (m), 1085 (m), 1052 (m), 969 (s) cm–1; 1H-NMR (C6D6) δ 5.29 (1 H, dt, J = 15.2, 6.7 Hz), 5.01 (1 H, ddt, J = 15.2, 8.8, 1.2 Hz), 3.85 (1 H, ddd, J = 11.6, 8.0, 3.2 Hz), 3.73 (1 H, dt, J = 11.6, 6.4 Hz), 2.85 (1 H, dd, J = 8.0, 6.4 Hz), 2.15 (1 H, app. d, J = 13.4 Hz), 2.04 (1 H, dddd, J = 11.7, 11.5, 8.8, 3.7 Hz), 1.86 (3 H, m), 1.75 (1 H, tdd, J = 13.4, 6.0, 1.0 Hz), 1.45 (2 H, m), 1.22 (4 H, m), 1.19 (1 H, tt, J = 13.5, 3.7 Hz), 1.11 (1 H, tdd, J = 13.4, 11.5, 3.7 Hz), 0.85 (3 H, t, J = 7.1 Hz) ppm; 13C-NMR (C6D6) δ 212.7 (C=O), 132.4 (=CH), 131.8 (=CH), 60.6 (CH2), 56.5 (CH), 45.0 (CH), 41.9 (CH2), 32.6 (CH2), 32.4 (CH2), 31.8 (CH2), 25.7 (CH2), 22.5 (CH2), 14.1 (CH3) ppm; MS m/z (%) 210 (M+, 3), 192 (8), 179 (100), 163 (6), 149 (13), 135 (26), 123 (54), 110 (33), 97 (21), 79 (81), 67 (97), 55 (87), 41 (99).
1b: Rf (isooctane/EtOAc, 1:1) 0.28; [α]Drt +7.0 (c 0.9, CHCl3); IR ν 3408 (br, m), 2956 (vs), 2930 (vs), 2873 (vs), 1707 (vs), 1460 (m), 1379 (m), 1312 (m), 1238 (w), 1138 (m), 1025 (s), 969 (s) cm–1; 1H-NMR (C6D6) δ 5.26 (1 H, dt, J = 15.2, 6.4 Hz), 5.17 (1 H, dd, J = 15.2, 8.9 Hz), 4.00 (1 H, ddd, J = 11.5, 8.8, 2.1 Hz), 3.34 (1 H, ddd, J = 11.5, 8.5, 5.3 Hz), 2.42 (1 H, dq, J = 8.9, 3.6 Hz), 2.36 (1 H, dtd, J = 8.8, 5.3, 1.0 Hz), 2.17 (1 H, app. d, J = 13.8 Hz), 1.99 (1 H, app. d, J = 5.3 Hz), 1.82 (3 H, m), 1.58 (1 H, m), 1.30 (3 H, m), 1.19 (4 H, m), 0.83 (3 H, t, J = 7.0 Hz) ppm; 13C-NMR (C6D6) δ 212.0 (C=O), 133.1 (=CH), 128.4 (=CH), 61.5 (CH2), 55.8 (CH), 43.6 (CH), 41.7 (CH2), 32.4 (CH2), 31.8 (CH2), 31.7 (CH2), 22.9 (CH2), 22.4 (CH2), 14.0 (CH3) ppm; MS m/z (%) 210 (M+, 2), 192 (10), 179 (14), 163 (6), 149 (9), 135 (32), 123 (50), 110 (98), 107 (29), 97 (47), 86 (48), 79 (83), 67 (100), 55 (82), 41 (98).
((1R,2S)-2-((E)-Hex-1-enyl)-6-oxocyclohexyl)methyl methanesulfonate (2a) and ((1S,2S)-2-((E)-hex-1-enyl)-6-oxocyclohexyl)methyl methanesulfonate (2b)
To an ice-cold solution of alcohol 1a (458 mg, 2.18 mmol) and Et3N (0.61 mL, 4.36 mmol) in CH2Cl2 (22 mL) was added MsCl (0.25 mL, 3.27 mmol). After stirring for 0.5 h, the reaction mixture was quenched with an aqueous saturated NaHCO3 solution and the product was extracted with CH2Cl2. The combined organic extracts were consecutively washed with a HCl solution (0.5 M; 5 mL) and a saturated NaHCO3 solution (8 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure to give mesylate 2a (604 mg, 96%). The same procedure applied to alcohol 1b afforded mesylate 2b as a solid in 99% yield.
2a: Rf (isooctane/EtOAc, 6:4) 0.37; [α]Drt –17.0 (c 1.0, CH3OH); IR ν 2956 (s), 2931 (s), 2860 (m), 1715 (vs), 1458 (m), 1356 (vs), 1251 (vw), 1174 (vs), 974 (s), 950 (s), 823 (w), 529 (s) cm–1; 1H-NMR (C6D6) δ 5.37 (1 H, dt, J = 15.3, 6.6 Hz), 4.95 (1 H, dd, J = 15.3, 9.0 Hz), 4.32 (1 H, dd, J = 9.5, 1.9 Hz), 4.21 (1 H, dd, J = 9.5, 5.7 Hz), 2.49 (3 H, s), 2.15 (1 H, app. d, J = 13.9 Hz), 2.02 (1 H, tdd, J = 11.8, 9.0, 3.7 Hz), 1.90 (2 H, m), 1.75 (1 H, ddd, J = 11.8, 5.7, 1.9), 1.69 (1 H, td, J = 13.9, 6.0 Hz), 1.42 (2 H, m), 1.27 (4 H, m), 1.15 (1 H, qt, J = 14.2, 3.5 Hz), 1.02 (1 H, qd, J = 12.9, 3.3 Hz), 0.91 (3 H, t, J = 7.3 Hz) ppm; 13C-NMR (C6D6) δ 206.5 (C=O), 132.9 (=CH), 131.0 (=CH), 66.0 (OCH2), 53.4 (CH), 44.4 (CH), 41.2 (CH2), 36.3 (SCH3), 32.4 (CH2), 32.2 (CH2), 31.6 (CH2), 25.1 (CH2), 22.4 (CH2), 14.0 (CH3) ppm; MS m/z (%) 289 (MH+, 8), 205 (4), 192 (79), 179 (17), 163 (34), 149 (36), 135 (65), 122 (66), 107 (36), 93 (43), 79 (100), 67 (42), 55 (46), 41 (47).
2b: mp 39 °C; Rf (isooctane/EtOAc, 6:4) 0.37; [α]Drt –28.5 (c 1.0, CH3OH); IR ν 2956 (s), 2932 (s), 2873 (m), 1711 (vs), 1459 (w), 1358 (vs), 1210 (vw), 1177 (vs), 1142 (vw), 959 (vs), 866 (w), 825 (w), 528 (m) cm–1; 1H-NMR (C6D6) δ 5.32 (1 H, dt, J = – 15.1, 6.8 Hz), 5.07 (1 H, dd, J = 15.1, 9.7 Hz), 4.51 (1 H, dd, J = 10.2, 6.5 Hz), 4.00 (1 H, dd, J = 10.2, 6.8 Hz), 2.61 (1 H, dq, J = 9.7, 3.4 Hz), 2.48 (1 H, app. q, J = 6.2 Hz), 2.26 (3 H, s), 2.11 (1 H, app. d, J = 13.2 Hz), 1.80 (2 H, m), 1.73 (1 H, td, J = 13.4, 6.0 Hz), 1.51 (1 H, m), 1.35 (3 H, m), 1.18 (4 H, m), 0.84 (3 H, t, J = 7.0 Hz) ppm; 13C-NMR (C6D6) δ 208.0 (C=O), 134.9 (=CH), 126.3 (=CH), 68.0 (OCH2), 52.4 (CH), 43.4 (CH), 41.5 (CH2), 36.2 (SCH3), 32.3 (CH2), 31.5 (CH2), 31.2 (CH2), 22.9 (CH2), 22.3 (CH2), 13.9 (CH3) ppm; MS m/z (%) 256 (5), 223 (4), 208 (14), 192 (24), 149 (24), 135 (16), 123 (14), 105 (16), 91 (35), 79 (56), 67 (51), 55 (76), 41 (100).
(2S,3S)-3-((E)-Hex-1-enyl)-2-methylcyclohexanol (3aa, 3ab) and (2R,3S)-3-((E)-hex-1-enyl)-2-methyl-cyclohexanol (3ba, 3bb)
LiAlH4 (143 mg, 3.76 mmol) in dry Et2O (7.5 mL) was refluxed for 30 min. Methanesulfonate 2a (546 mg, 1.89 mmol) in dry Et2O (7.5 mL) was dropwise added at reflux temperature over a period of 10 min. The reaction mixture was refluxed for 2 h, then cooled to 0 °C and quenched by the sequential addition of H2O (145 μL), NaOH (15% solution; 145 μL) and H2O (300 μL). After stirring at rt for 1h, the white precipitate was removed by filtration over Celite® and washed with EtOAc. The filtrate was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel (n-pentane/EtOAc, 9:1) to give alcohols 3aa (70 mg) and 3ab (285 mg) in 96% total yield. The same procedure applied to methanesulfonate 2b (1.06 g) afforded alcohols 3ba (141 mg) and 3bb (563 mg) in 98% total yield.
3aa: Rf (isooctane/EtOAc, 6:4) 0.54; [α]Drt –45.7 (c 1.0, CHCl3); IR ν 3392 (br, m), 2957 (s), 2928 (vs), 2872 (s), 1456 (m), 1376 (w), 1212 (w), 963 (w), 879 (w) cm–1; 1H-NMR (CDCl3) δ 5.37 (1 H, dt, J = 15.2, 6.2 Hz), 5.15 (1 H, ddt, J = 15.2, 8.6, 1.1), 3.88 (1 H, m, ΣJ = 25 Hz), 1.96 (3 H, m), 1.81 (1 H, app. d, J = 13.2 Hz), 1.64 (2 H, m), 1.46 (2 H, m), 1.35–1.24 (5 H, m), 1.11 (1 H, m), 0.92 (3 H, d, J = 7.0 Hz), 0.87 (3 H, t, J = 7.0 Hz) ppm; 13C-NMR (CDCl3) δ 134.7 (=CH), 130.1 (=CH), 71.1 (OCH), 41.7 (CH), 40.6 (CH), 33.3 (CH2), 33.3 (CH2), 32.3 (CH2), 31.9 (CH2), 22.2 (CH2), 19.6 (CH2), 16.7 (CH3), 14.0 (CH3) ppm; MS m/z (%) 196 (M+, <1), 178 (54), 163 (8), 149 (25), 135 (56), 121 (25), 111 (50), 93 (51), 79 (52), 67 (54), 55 (54), 41 (100).
3ab: Rf (isooctane/EtOAc, 6:4) 0.47; [α]Drt +12.3 (c 1.0, CHCl3); IR ν 3350 (br, m), 2958 (s), 2926 (vs), 2857 (s), 1458 (m), 1376 (w), 1358 (w), 1294 (vw), 1117 (vw), 1032 (s), 966 (vs), 858 (vw) cm–1; 1H-NMR (CDCl3) δ 5.37 (1 H, dt, J = 15.2, 6.7 Hz), 5.17 (1 H, ddd, J = 15.2, 8.5, 1.2), 3.15 (1 H, m, ΣJ = 39 Hz), 1.96 (3 H, m), 1.72 (1 H, app. d, J = 13.0 Hz), 1.57 (2 H, m), 1.43 (1 H, s), 1.37–1.20 (6 H, m), 1.10 (1 H, m), 1.06 (1 H, m), 0.97 (3 H, d, J = 6.6 Hz), 0.87 (3 H, t, J = 6.9 Hz) ppm; 13C-NMR (CDCl3) δ 134.0 (=CH), 130.4 (=CH), 76.1 (OCH), 47.3 (CH), 44.6 (CH), 35.5 (CH2), 33.4 (CH2), 32.2 (CH2), 31.8 (CH2), 23.9 (CH2), 22.2 (CH2), 16.0 (CH3), 14.0 (CH3) ppm; MS m/z (%) 196 (M+, <1), 178 (22), 163 (5), 149 (22), 135 (29), 121 (42), 108 (21), 93 (40), 79 (47), 67 (52), 55 (59), 41 (100).
3ba: Rf (isooctane/EtOAc, 8:2) 0.47; [α]Drt –40.3 (c 1.0, CHCl3); IR ν 3349 (br, m), 2956 (s), 2928 (vs), 2872 (s), 1460 (m), 1377 (w), 1142 (w), 1042 (m), 1017 (m), 967 (s) cm–1; 1H-NMR (CDCl3) δ 5.40 (2 H, m), 3.56 (1 H, td, J = 7.7, 3.8 Hz), 2.41 (1 H, m), 2.20 (2 H, m), 1.85 (1 H, m), 1.59 (3 H, m), 1.50 (2 H, m), 1.41–1.28 (5 H, m), 0.90 (3 H, d, J = 6.9 Hz), 0.89 (3 H, t, J = 6.5 Hz) ppm; 13C-NMR (CDCl3) δ 131.2 (=CH), 130.9 (=CH), 72.4 (CH), 41.8 (CH), 41.6 (CH), 32.7 (CH2), 32.5 (CH2), 31.9 (CH2), 29.6 (CH2), 22.2 (CH2), 20.4 (CH2), 14.5 (CH3), 14.0 (CH3) ppm; MS m/z (%) 196 (M+, 4), 178 (14), 163 (4), 149 (17), 135 (22), 121 (42), 111 (39), 93 (42), 79 (48), 67 (47), 55 (49), 41 (100).
3bb: Rf (isooctane/EtOAc, 8:2) 0.47; [α]Drt –10.3 (c 1.0, CHCl3); IR ν 3350 (br, m), 2928 (vs), 2860 (s), 1466 (m), 1447 (m), 1378 (w), 1342 (w), 1301 (w), 1119 (w), 1053 (m), 1016 (m), 968 (s) cm–1; 1H-NMR (CDCl3) δ 5.41 (1 H, dd, J = 15.5, 5.3 Hz), 5.38 (1 H, dt, J = 15.5, 5.8 Hz), 3.72 (1 H, dt, J = 10.6, 4.1 Hz), 2.11 (1 H, m), 1.97 (3 H, m), 1.72 (1 H, m), 1.59 (1 H, app. d, J = 12.3 Hz), 1.42 (1 H, m); 1.37–1.20 (7 H, m), 0.87 (3 H, t, J = 6.8 Hz), 0.80 (3 H, d, J = 7.0 Hz) ppm; 13C-NMR (CDCl3) δ 133.3 (=CH), 129.5 (=CH), 73.6 (CH), 42.8 (CH), 39.8 (CH), 32.4 (CH2), 31.9 (CH2), 29.0 (CH2), 24.8 (CH2), 23.2 (CH2), 22.2 (CH2), 14.0 (CH3), 6.7 (CH3) ppm; MS m/z (%) 196 (M+,<1), 178 (40), 163 (6), 149 (22), 135 (32), 122 (28), 111 (36), 108 (25), 93 (36), 79 (50), 67 (34), 55 (51), 41 (100).
(2S,3S)-3-((E)-Hex-1-enyl)-2-methylcyclohexanone (4a) and (2R,3S)-3-((E)-hex-1-enyl)-2-methyl-cyclohexanone (4b)
To a solution of (COCl)2 (113 μL, 1.33 mmol) in dry CH2Cl2 (4 mL) was dropwise added a solution of dimethyl sulfoxide (DMSO; 189 μL, 2.66 mmol) in dry CH2Cl2 (750 μL) at –78 °C. After 2 minutes of stirring, a mixture of alcohols 3aa and 3ab (238 mg, 1.21 mmol) in dry CH2Cl2 (3 mL) was dropwise added over a period of 5 min and the mixture was stirred for 15 min. Et3N (845 μL, 6.66 mmol) was then added and the reaction mixture was stirred for an additional 5 min, followed by slow warming to rt. H2O (10 mL) was added and the product was extracted with CH2Cl2. The combined organic layers were concentrated under reduced pressure and the residue was diluted with t-butyl methyl ether (MTBE). After washing with H2O, the organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (n-pentane/EtOAc, 98:2) to give cyclohexanone 4a (209 mg, 89%). The same procedure applied to the mixture of alcohols 3ba and 3bb afforded cyclohexanone 4b in 96% yield.
4a: Rf (isooctane/EtOAc, 8:2) 0.44; [α]Drt –12.0 (c 1.0, CH3OH); IR ν 2930 (vs), 2863 (vs), 1712 (vs), 1450 (m), 1376 (w), 1329 (w), 1307 (w), 1216 (w), 1181 (w), 1128 (bw), 1017 (w), 969 (s) cm–1; 1H-NMR (C6D6) δ 5.24 (1 H, dt, J = 15.2, 6.6 Hz), 5.05 (1 H, dd, J = 15.2, 8.4 Hz), 2.26 (1 H, app. d, J = 13.4 Hz), 1.92 (2 H, m), 1.86 (1 H, td, J = 13.6, 6.0 Hz), 1.77 (1 H, m), 1.73 (1 H, m), 1.56 (1 H, m), 1.48 (1 H, app. d, J = 13.0 Hz), 1.32 (1 H, m), 1.26 (4 H, m), 1.17 (1 H, m), 1.11 (3 H, d, J = 6.0 Hz), 0.86 (3H, t, J = 7.2 Hz) ppm; 13C-NMR (C6D6) δ 209.5 (C=O), 133.4 (=CH), 130.9 (=CH), 50.4 (CH), 49.4 (CH), 41.5 (CH2), 32.9 (CH2), 32.3 (CH2), 31.1 (CH2), 26.0 (CH2), 21.6 (CH2), 14.1 (CH3), 13.0 (CH3) ppm; MS m/z (%) 194 (M+, 9), 137 (4), 123 (30), 110 (19), 96 (4), 81 (45), 79 (32), 67 (100), 47 (67).
4b: Rf (isooctane/EtOAc, 8:2) 0.44; [α]Drt –13.1 (c 1.0, CHCl3); IR ν 2957 (s), 2930 (vs), 2872 (s), 1713 (vs), 1448 (m), 1377 (vw), 1312 (vw), 1222 (w), 1141 (w), 1078 (w), 995 (w), 968 (m) cm–1; 1H-NMR (C6D6) δ 5.32 (1 H, dtd, J = 15.3, 6.8, 1.1 Hz), 5.20 (1 H, ddd, J = 15.3, 8.8, 0.9 Hz), 2.36 (1 H, m), 2.20 (2 H, m), 1.87 (3 H, m), 1.65 (1 H, m), 1.44 (3 H, m), 1.22 (4 H, m), 0.97 (3 H, dd, J = 6.8, 1.3 Hz), 0.83 (3 H, t, J = 7.2 Hz) ppm; 13C-NMR (C6D6) δ 210.6 (C=O), 133.1 (=CH), 129.0 (=CH), 48.5 (CH), 47.0 (CH), 41.0 (CH2), 32.6 (CH2), 32.0 (CH2), 31.1 (CH2), 23.5 (CH2), 22.5 (CH2), 14.1 (CH3), 12.9 (CH3) ppm; MS m/z (%) 194 (M+, 8), 179 (2), 149 (5), 138 (3), 123 (34), 110 (22), 95 (14), 81 (57), 67 (67), 55 (49), 49 (100), 41 (93).

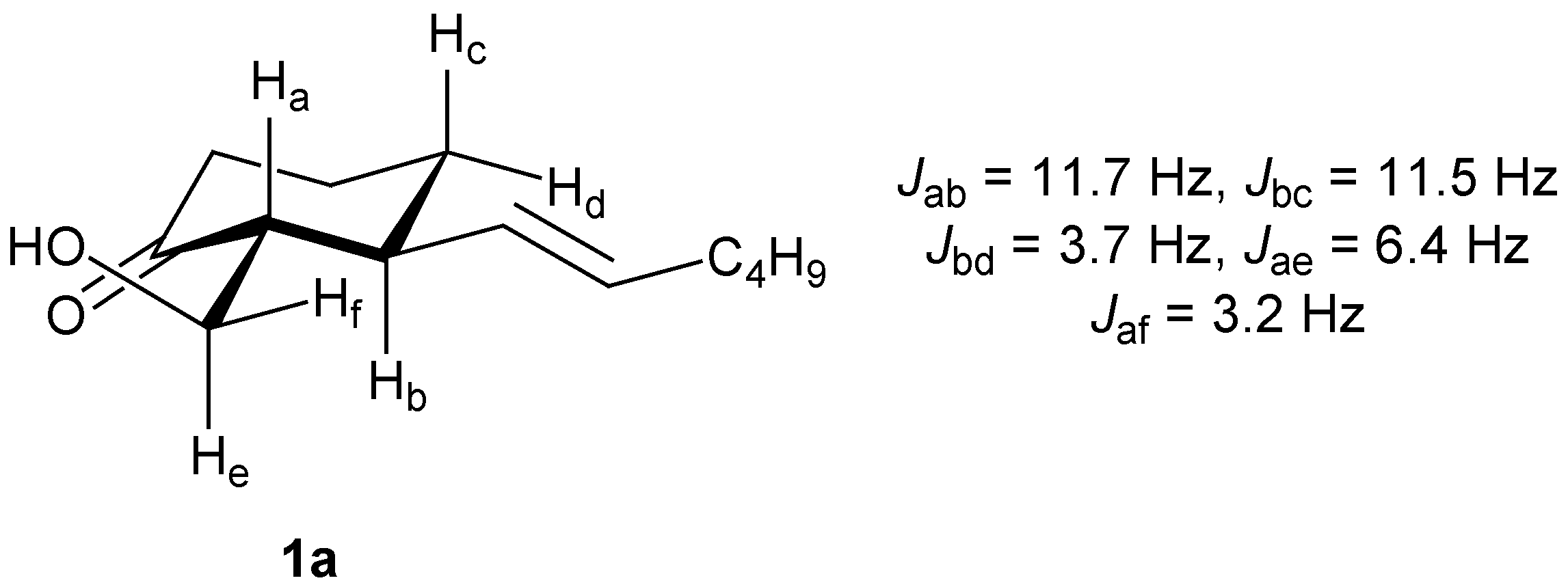{kind=link}
{kind=link}
{kind=link}


