Results and Discussion
The synthesis of the new di-(
β-chloroethyl)amides with their active group supported by
N-(
meta-acylaminobenzoyl)-
D,L-aminoacids was performed in several steps. First the
N-(
meta-acylamino-benzoyl)-
L-aminoacids
4-9 were obtained by reaction of the previously obtained
N-(
meta-aminobenzoyl)-
L-aminoacids
1-3 [
15,
16] with formic acid when N-(
meta-formylaminobenzoyl)-
L-aminoacids
4-6 compounds have been obtained or acetic anhydride in acetic acid medium when N-(
meta-acetylaminobenzoyl)-
L-aminoacids
7-9 compounds have been obtained (
Scheme 1).
Scheme 1.
Synthesis of the N-(meta-acylaminobenzoyl)-L-aminoacids 4-9.
Scheme 1.
Synthesis of the N-(meta-acylaminobenzoyl)-L-aminoacids 4-9.
The six N-acyl derivatives which had not been previously decribed in the literature were obtained as crystalline products melting within a narrow temperature range. The structures of these compounds were ascertained by means of elemental analyses and IR and 1H-NMR spectral measurements. Absorption bands characteristic of these structures were noticed in their IR spectra, namely the amide I and amide II bands within 1635-1710 cm-1 and 1520-1583 cm-1, respectively. Every compound showed an absorption band characteristic of the meta-substituted benzene ring within the 685-830 cm-1 range. In addition to that, the compound 6 and 9 also presented bands characteristic of the C-S valence bands at 728-754 cm-1. The signal at 5.50 ppm in the 1H-NMR of the compounds 1-3, was also presented in the spectra of the reaction products 4-9 between 5.70-6.30 ppm due to the conjugation effects with the >C=O group.
By heating the N-(
meta-acylaminobenzoyl)-
L-aminoacids
4-9 with acetic anhydride at 100°C the ring closure at the
α-carboxyl group takes place, which results in the formation of Δ
2-oxazolin-5-ones
10-15 (
Scheme 2).
Scheme 2.
Synthesis of the Δ2-oxazolin-5-ones 10-15.
Scheme 2.
Synthesis of the Δ2-oxazolin-5-ones 10-15.
The cyclization reaction may be considered to take place according to a mechanism similar to that described in literature for other 2,4-disubstituted-Δ
2-oxazolones [
11,
15,
17,
18,
19]. The products
10-15 were obtained in 62-99% yields as crystalline compounds with sharp melting points. The oxazolone structures were elucidated by means of elemental analysis data and IR and
1H-NMR spectral measurements. The IR absorption bands characteristic of the >C=O group are to be found at 1630-1820 cm
-1. The NH group vibrations give a single wide band within the 3080-3500 cm
-1 range. The C=N bond is made evident by an absorption maximum at 1610-1625 cm
-1 and the C-S bond at 654-725 cm
-1. The
1H-NMR spectra are indicative of the cyclization realization by the disappearance of the signal characteristic of the acylated amine group at 5.50-6.50 ppm as well as by the heterocyclic proton descreening in virtue of its neighborhoods.
The high reactivity of Δ
2-oxazolin-5-ones towards numerous nucleophilic reagents [
11,
17,
18,
20] prompted us to perform the reaction between the newly obtained oxazolones
10-15 and di-(
β-chloroethyl)amine to obtain new
N-mustards
16-21 with possible higher activity and lower toxicity; the electron of the nitrogen atom being involved in an amidic conjugation system (
Scheme 3).
The
N-mustards are solid compounds with fixed melting points and stable to long preservation in a moistureless medium. Their structures were elucidated by means of elemental analyses and spectral measurements (IR,
1H-NMR). The IR spectra of the compounds
16-21 show the amide I and II bands as well as intense absorption maxima within the 670-800 cm
-1 range corresponding to the C-Cl valence bond. The spectra of the compounds
18 and
21 show an additional band at 718-771 cm
-1 attributable to the C-S group. The
1H-NMR spectra also reveal the oxazolone ring opening. Some spectra of the synthesized compounds are presented in
Figure 1,
Figure 2,
Figure 3 and
Figure 4.
Scheme 3.
Synthesis of the N-mustards 16-21.
Scheme 3.
Synthesis of the N-mustards 16-21.
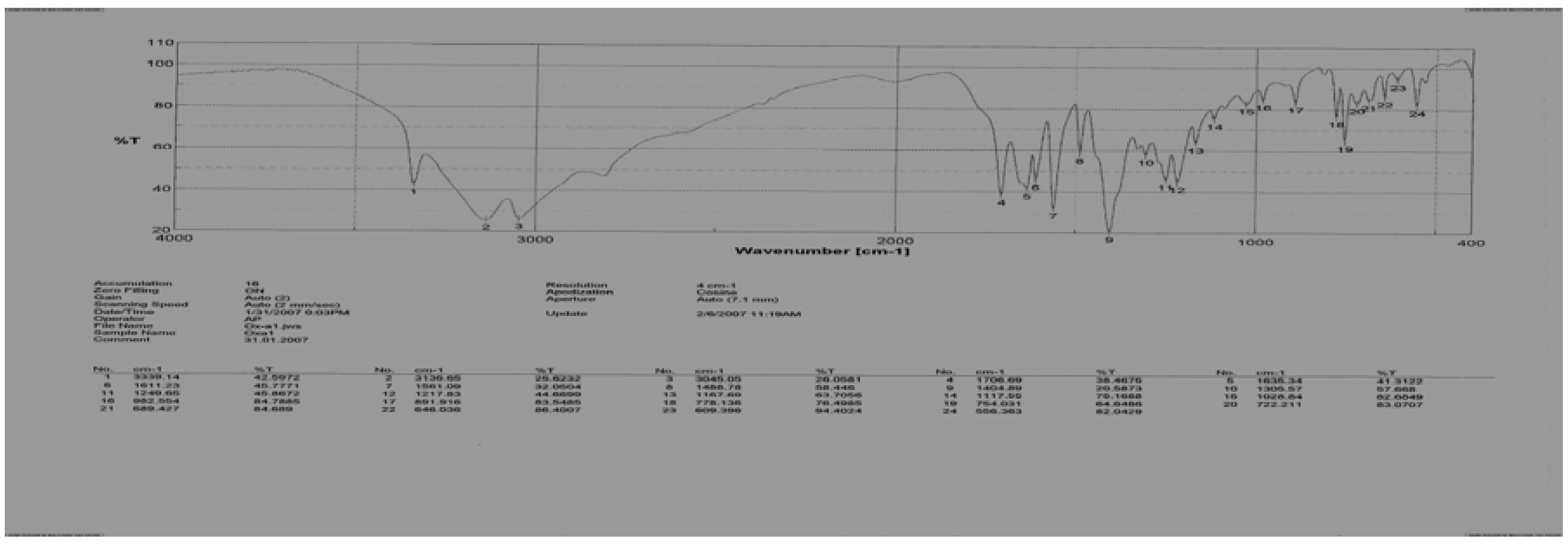
Figure 1.
IR spectrum of the N-(meta-formylaminobenzoyl)-L-methionine (6)
Figure 1.
IR spectrum of the N-(meta-formylaminobenzoyl)-L-methionine (6)
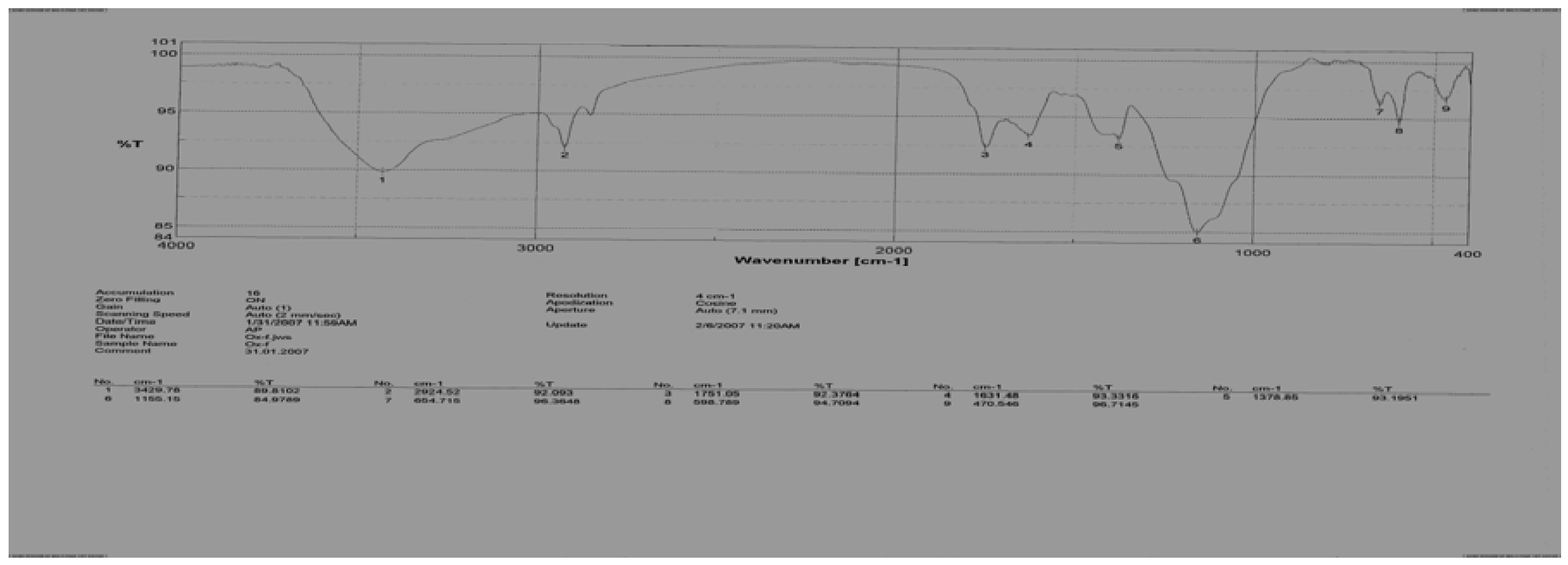
Figure 2.
IR spectrum of 2-(meta-formylaminophenyl)-4-(β-methylthio-ethyl)-Δ2-oxazolin-5-one (12).
Figure 2.
IR spectrum of 2-(meta-formylaminophenyl)-4-(β-methylthio-ethyl)-Δ2-oxazolin-5-one (12).
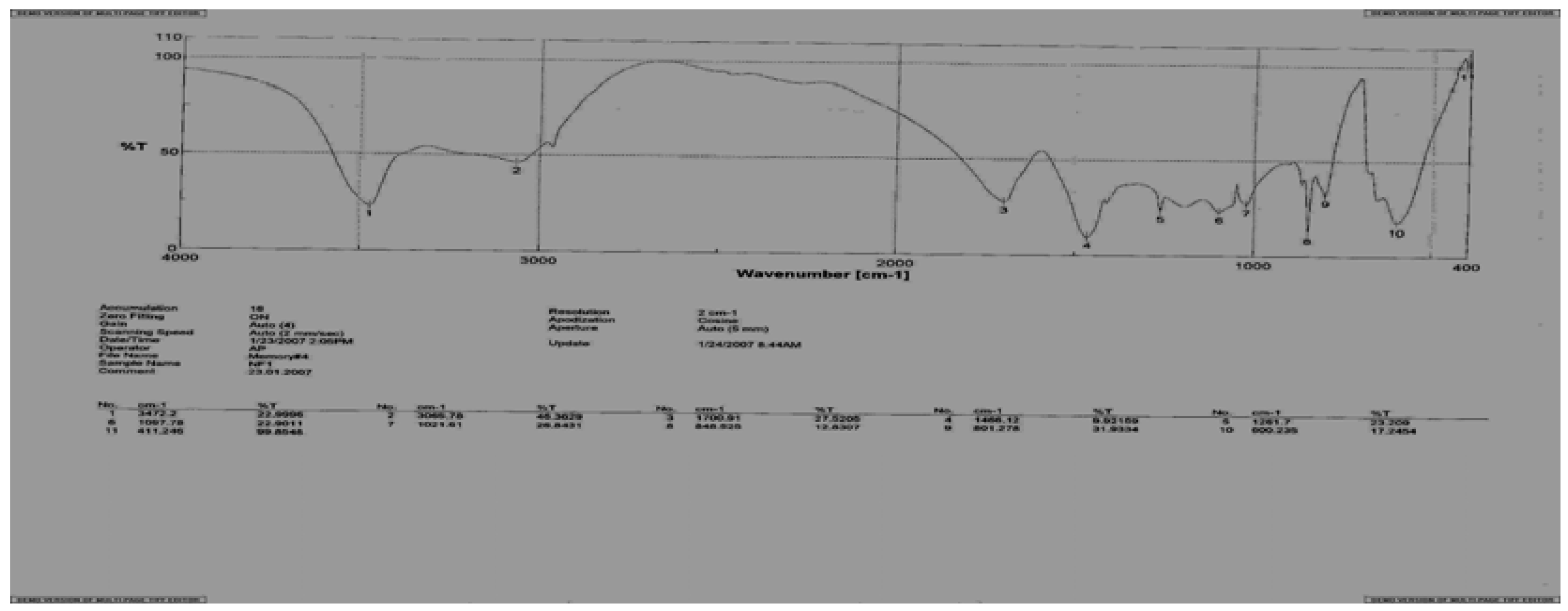
Figure 3.
IR spectrum of the α-[N-di-(β-chloroethyl)-amide] of N'-(meta-formylaminobenzoyl)-D,L-methionine (18).
Figure 3.
IR spectrum of the α-[N-di-(β-chloroethyl)-amide] of N'-(meta-formylaminobenzoyl)-D,L-methionine (18).
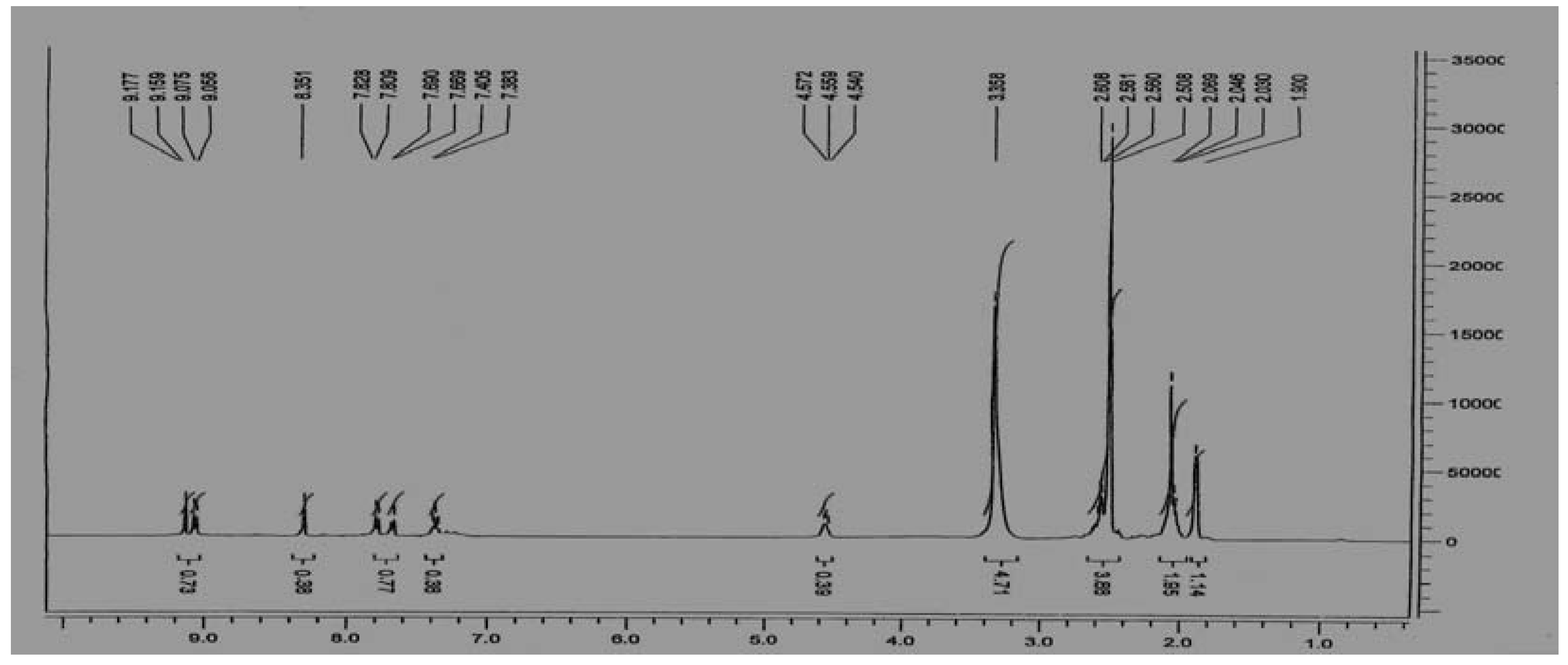
Figure 4.
1H-NMR spectrum of the α-[N-di-(β-chloroethyl)-amide] of N'-(meta-acetylaminobenzoyl)-D,L-methionine (21).
Figure 4.
1H-NMR spectrum of the α-[N-di-(β-chloroethyl)-amide] of N'-(meta-acetylaminobenzoyl)-D,L-methionine (21).
Biological activity
The six synthesized
N-mustards
16-21 were tested for their toxicity (DL
50 – median lethal dose) and an experimental study on their antitumoural activity was performed. The antitumor activity was studied using two experimental tumours frequently used in experiments with functionalized aminoacids: Ehrlich ascite and Walker 256 carcinosarcoma. The data in
Table 1 reveal the inhibitory effect of the synthesized
N-mustards on the experimental tumors. At the same time the inhibition values on the liquid tumors were noticed to be higher than on the solid ones, meaning that the Ehrlich ascite is more sensitive to the action of the tested substances. The compounds
16-21 containing a
p‑acetylamino moiety in their structure show the highest inhibition values, especially on the liquid tumor. The di-(
β-chloroethyl)amino group grafted on
N-(
meta-aminobenzoyl)-aminoacids
1-3 is seen to be much less toxic and the obtained di-(
β-chloroethyl)-amides
16-21 are selective enough toward the neoplastic cells.
Table 1.
DL50 values and inhibition (%) induced by the N-mustards 16-21 on some experimental tumors.
Table 1.
DL50 values and inhibition (%) induced by the N-mustards 16-21 on some experimental tumors.
| Compound | DL50 mg/Kg body | Inhibition (%) |
|---|
| Ehrlich ascite | Walker 256 carcinosarcoma |
|---|
| mg/Kg body | - | 400 | 200 | 40 | 400 | 200 | 40 |
| 16 | 770 | 35 | 32 | 28 | 27 | 21 | 18 |
| 17 | 810 | 42 | 38 | 32 | 36 | 30 | 23 |
| 18 | 898 | 46 | 41 | 35 | 40 | 36 | 31 |
| 19 | 986 | 56 | 49 | 42 | 48 | 42 | 35 |
| 20 | 1020 | 54 | 48 | 41 | 51 | 46 | 40 |
| 21 | 1270 | 58 | 52 | 47 | 53 | 47 | 41 |
| Sarcolisine | 223 | 56 | 52 | 48 | 50 | 48 | 40 |
In comparison with sarcolisine, a well-known antitumoural agent, the compounds
16-21 showed a similar inhibition on both Ehrlich ascite and Walker 256 carcinosarcoma. The great advantage of the tested compounds consists in their much lower toxicity (DL
50 values) than that of sarcolisine (
Table 1). This could be explained by the presence of the peptide support involved in the cytotoxic group transport to the malignant cell level where it would inhibit the abnormal synthesis of some proteins and nucleoproteids by an alkylating mechanism. The cytostatic action of di-(
β-chloroethyl)amines may be assumed to consist in the possibility of an intramolecular cyclization in aqueous solution affording the formation of an immonium cation consisting of a three-membered ethyleneiminic cation ring which is unstable and can be easily broken being thus able to act by means of the
β-carbon atom as an alkylating reagent for the substances containing active groups such as –NH, -SH, etc. (
Scheme 4).
Scheme 4.
The alkylating mechanism of the di-(β-chloroethyl)-amines.
Scheme 4.
The alkylating mechanism of the di-(β-chloroethyl)-amines.
The alkylating property of di-(β-chloroethyl)amines allow us to assume that the N-mustards 16-21 are linked by means of the di-(β-chloroethyl)-amino group to the active groups –NH, -SH etc. of proteins, nucleoproteids or desoxyribonucleic acid molecules which participate to the cell multiplication process, and thus the anarchical division of cancerous cells is blocked.
Experimental Section
General
All melting points were determined on a Melt-Temp R apparatus equipped with a digital thermometer and are uncorrected. C,H,N analyses were performed on an Elemental Exeter Analytical CE 440 Apparatus. The IR spectra were recorded with KBr pellets on a Digilab Scimitar Series spectrophotometer. The 1H-NMR spectra were recorded on Bruker ARX-300 (300 MHz) or ARX-400 (400 MHz) spectrometers. Chemical shifts were recorded as δ values in parts per million (ppm). All chemical reagents were obtained from Aldrich Chemical Company.
General synthesis method of N-meta-(formylaminobenzoyl) aminoacids 4-6
N-(meta-aminobenzoyl)-L-aminoacid 1-3, (0.02 mol) was introduced in a reaction flask provided with a refluxing condenser, then 80% formic acid (10 mL) was added and the mixture heated on an oil bath for 30 minutes, at 115-120°C. After cooling, anhydrous acetone (20 mL) was added dropwise under stirring, when colorless crystalline products separated. The product was then filtered, dried in a vacuum oven at 45°C and finally recrystallized from an ethyl alcohol–acetone mixture.
N-(meta-Formylaminobenzoyl)-L-asparagic acid (4).White solid; yield 88 % (491 mg); m.p. 76-78°C. Anal. Calc. for C12H12N2O6: 51.42 % C, 4.32 % H, 10.00 % N; Found: 51.88 % C, 4.56 % H, 10.33 % N; IR (ν, cm-1): 2500, 3340 (NH); 1525, 1670 (CO); 685 (CH); 1H-NMR δ (CD3CN): 2.30 (d, 2H, CH2); 4.95 (t, 1H, CH); 6.20 (s, 1H, NH); 7.65 (t, 1H, Ar); 7.80 (d, 1H, Ar); 8.05 (d, 1H, Ar); 8.10 (s, 1H, Ar).
N-(meta-Formylaminobenzoyl)-L-asparagine (5). White solid; yield 78 % (434 mg); m.p. 156-158°C. Anal. Calc. for C12H13N3O5: 51.60 % C, 4.69 % H, 15.05 % N; Found: 51.38 % C, 5.06 % H, 15.33 % N; IR (ν, cm-1): 2500, 3200 (NH); 1535, 1710 (CO); 685 (CH); 1H-NMR δ (CF3COOD, 300 MHz): 2.25 (d, 2H, CH2); 4.90 (t, 1H, CH); 6.00 (s, 1H, NH); 7.60 (t, 1H, Ar); 7.85 (d, 1H, Ar); 8.0 (d, 1H, Ar); 8.15 (s, 1H, Ar).
N-(meta-Formylaminobenzoyl)-L-methionine (6). White solid; yield 72 % (426 mg); m.p. 195-196°C. Anal. Calc. for C13H16N2O4S: 52.70 % C, 5.40 % H, 9.45 % N, 10.81 % S; Found: 52.68 % C, 5.23 % H, 9.31 % N, 10.58 % S; IR (ν, cm-1): 3339, 3136 (NH); 1706, 1635 (CO); 828 (CH); 754 (C-S); 1H-NMR δ (DMSO-d6, 400 MHz): 2.08 (s, 3H, CH3); 2.50 (m, 4H, CH2); 4.57 (t, 1H, CH); 6.26 (d, 1H, NH); 7.80 (t, 1H, Ar); 8.34 (d, 1H, Ar); 8.74 (s, 1H, Ar); 9.06 (d, 1H, NH); 9.80 (d, 1H, CH).
General synthesis method of N-(meta-acetylaminobenzoyl)-aminoacids 7-9
A reaction mixture consisting of N-(meta-aminobenzoyl)-L-amino acid 1-3, (0.02 mol), 96% acetic acid (10 mL) and acetic anhydride (4 mL) was refluxed for 4 hours in a reaction flask provided with a refluxing condenser. The reaction mixture was the passed into a crystallizer and allowed to stand for 24 hours at room temperature, when the crystalline products separated. It was filtered off, washed two-three times with acetone (5 mL each portion), dried in a vacuum oven at 45°C and finally purified by recrystallization from boiling ethyl alcohol.
N-(meta-Acetylaminobenzoyl)-L-asparagic acid (7). White solid; yield 69.5 % (398 mg); m.p. 120-122°C. Anal. Calc. for C13H14N2O6: 53.05 % C, 4.80 % H, 9.52 % N; Found: 53.54 % C, 5.03 % H, 9.86 % N; IR (ν, cm-1): 2500, 3300 (NH); 1530, 1700 (CO); 690 (CH); 1H-NMR δ (CD3COCD3, 300 MHz): 2.25 (d, 2H, CH2); 4.50 (s, 3H, CH3); 5.05 (t, 1H, CH); 5.70 (s, 1H, NH); 7.70 (t, 1H, Ar); 7.85 (d, 1H, Ar); 8.00 (d, 1H, Ar).
N-(meta-Acetylaminobenzoyl)-L-asparagine (8). White solid; yield 73.8 % (432 mg); m.p. 175-177°C. Anal. Calc. for C13H15N3O5: 53.22 % C, 5.16 % H, 14.33 % N; Found: 53.44 % C, 5.63 % H, 14.68 % N; IR (ν, cm-1): 2600, 3300 (NH); 1520, 1680 (CO); 695 (CH); 1H-NMR δ (CD3COCD3, 300 MHz): 2.30 (d, 2H, CH2); 4.50 (s, 3H, CH3); 5.00 (t, 1H, CH); 5.80 (s, 1H, NH); 7.75 (t, 1H, Ar); 7.90 (d, 1H, Ar); 8.05 (d, 1H, Ar); 8.25 (s, 1H, Ar).
N-(meta-Acetylaminobenzoyl)-L-methionine (9). Grey solid; yield 70.32 % (432 mg); m.p. 272-273°C. Anal. Calc. for C14H18N2O4S: 54.19 % C, 5.80 % H, 9.03 % N, 10.32 % S; Found: 53.98 % C, 5.63 % H, 9.07 % N, 10.18 % S; IR (ν, cm-1): 2800, 3304 (NH); 1583, 1684 (CO); 830 (CH); 728 (C-S); 1H-NMR δ (DMSO-d6, 400 MHz): 1.90 (s, 3H, CH3); 2.06 (s, 3H, CH3); 2.12-2.60 (m, 4H, CH2); 4.55 (t, 1H, CH); 7.38 (t, 1H, Ar); 7.66 (d, 1H, Ar); 7.80 (d, 1H, Ar); 8.35 (s, 1H, Ar); 9.05 (d, 1H, NH); 9.15 (s, 1H, NH).
General synthesis method of 2-(meta-acylaminophenyl)-4-(β-carboxymethyl-, β-amidomethyl-, β-methyl-thioethyl)-Δ2-oxazolin-5-ones 10-15
N-(meta-acylaminobenzoyl)-L-amino acid 4-9 (0.02 mol) and acetic anhydride (14 mL) were introduced in a flask provided with a reflux condenser and heated on an oil bath for 60 minutes at 140 °C. After cooling, a thin stream of the resulting solution was poured under stirring into a mixture of anhydrous ethyl ether (40 mL) and anhydrous petroleum ether (50 mL). The solvent mixture was then decanted and the remaining viscous mass washed repeatedly with anhydrous ethyl ether till it turned into a fine powder. It was filtered off, repeatedly washed on the filter with anhydrous ethyl ether and allowed to dry in a vacuum oven for 8 hours at 40-45°C. Finally it was purified by recrystallization from anhydrous dioxane.
2-(meta-Formylaminophenyl)-4-(β-carboxymethyl)-Δ2-oxazolin-5-one (10). Yellow solid; yield 67 % (351 mg); m.p. 180-182°C. Anal. Calc. for C12H10N2O5: 54.97 % C, 3.84 % H, 10.68 % N; Found: 54.08 % C, 4.24 % H, 10.12 % N; IR (ν, cm-1): 3320, 3520 (NH); 1775, 1800 (CO); 1080 (C-O-C); 1620 (C=N); 710 (CH); 1H-NMR δ (CD3CN, 300 MHz): 2.90-3.00 (m, 2H, CH2); 4.95 (t, 1H, CH); 6.60 (s, 1H, NH); 7.65 (t, 1H, Ar); 7.95 (d, 2H, Ar); 8.20 (s, 1H, Ar).
2-(meta-Formylaminophenyl)-4-(β-amidomethyl)-Δ2-oxazolin-5-one (11). Yellow solid; yield 69 % (360 mg); m.p. 198-200°C. Anal. Calc. for C12H10N3O4: 55.17 % C, 4.24 % H, 16.09 % N; Found: 55.41 % C, 4.54 % H, 16.61 % N; IR (ν, cm-1): 3325, 3500 (NH); 1760, 1795 (CO); 1088 (C-O-C); 1625 (C=N); 715 (CH); 1H-NMR δ (DMSO-d6, 300 MHz): 2.90-3.00 (m, 2H, CH2); 5.00 (t, 1H, CH); 6.60 (s, 2H, NH2); 7.70 (t, 1H, Ar); 7.95 (d, 2H, Ar); 8.20 (s, 1H, Ar).
2-(meta-Formylaminophenyl)-4-(β-methylthioethyl)-Δ2-oxazolin-5-one (12). Yellow solid; yield 62.2 % (345 mg); m.p. 182-183°C. Anal. Calc. for C13H14N2O3S: 56.11 % C, 5.03 % H, 10.07 % N, 11.51 % S; Found: 55.83 % C, 4.87 % H, 10.01 % N, 11.32 % S; IR (ν, cm-1): 3429 (NH); 1751, 1631 (CO); 1610 (C=N); 1155 (C-O-C); 835 (CH); 654 (C-S); 1H-NMR δ (DMSO-d6, 300 MHz): 2.08 (s, 3H, CH3); 2.50 (m, 4H, CH2); 4.57 (t, 1H, CH); 6.22 (d, 1H, NH); 7.80 (t, 1H, Ar); 8.32 (d, 2H, Ar); 8.39 (d, 1H, Ar); 8.74 (s, 1H, Ar); 9.70 (d, 1H, CH).
2-(meta-Acetylaminophenyl)-4-(β-carboxymethyl)-Δ2-oxazolin-5-one (13). Yellow solid; yield 63.8 % (352 mg); m.p. 149-150°C. Anal. Calc. for C13H12N2O5: 56.52 % C, 4.38 % H, 10.14 % N; Found: 56.98 % C, 4.74 % H, 10.62 % N; IR (ν, cm-1): 3084, 3210 (NH); 1730, 1770 (CO); 1084 (C-O-C); 1620 (C=N); 728 (CH); 1H-NMR δ (CD3CN, 300 MHz): 2.95 (d, 2H, CH2); 4.50 (s, 3H, CH3); 5.20 (t, 1H, CH); 6.50 (s, 2H, NH); 7.70 (t, 1H, Ar); 8.00 (d, 2H, Ar); 8.25 (s, 1H, Ar).
2-(meta-Acetylaminophenyl)-4-(β-amidoethyl)-Δ2-oxazolin-5-one (14). White solid; yield 61.5 % (337 mg); m.p. 168-170°C. Anal. Calc. for C13H13N3O4: 56.72 % C, 4.76 % H, 15.27 % N; Found: 56.91 % C, 4.95 % H, 15.43 % N; IR (ν, cm-1): 3100, 3320 (NH); 1770, 1800 (CO); 1084 (C-O-C); 1620 (C=N); 705 (CH); 1H-NMR δ (DMSO-d6, 300 MHz): 2.80 (d, 2H, CH2); 4.50 (s, 3H, CH3); 5.05 (t, 1H, CH); 5.70 (s, 1H, NH); 6.60 (s, 2H, NH2); 7.70 (t, 1H, Ar); 7.90 (d, 2H, Ar); 8.20 (s, 1H, Ar).
2-(meta-Acetylaminophenyl)-4-(β-methylthioethyl)-Δ2-oxazolin-5-one (15). Brown solid; yield 58.7 % (342 mg); m.p. 142-143°C. Anal. Calc. for C14H16N2O3S: 57.53 % C, 5.47 % H, 9.58 % N, 10.95 % S; Found: 57.18 % C, 5.21 % H, 9.24 % N, 10.81 % S; IR (ν, cm-1): 3080, 3240 (NH); 1680, 1815 (CO); 1078 (C-O-C); 1615 (C=N); 820 (CH); 725 (C-S); 1H-NMR δ (DMSO-d6, 400 MHz): 2.07 (s, 3H, CH3); 2.26 (s, 3H, CH3); 2.57 (m, 4H, CH2); 4.57 (m, 1H, CH); 5.73 (s, 1H, NH); 7.80 (t, 1H, Ar); 8.33 (d, 1H, Ar); 8.39 (d, 1H, Ar); 8.74 (s, 1H, Ar).
General synthesis of α-[N-di-(β-chloroethyl)amides] of the N’-(meta-acylaminobenzoyl)-D,L-amino-acids 16-21
Δ2-Oxazolin-5-ones 10-15 (0.02 mol), di-(β-chloroethyl)amine hydrochloride (0.004 mol), anhydrous dioxane (20 mL) and triethylamine (0.556 mL) were introduced in a flask provided with a refluxing condenser, heated at 50-60°C for one hour and the resulting triethylamine hydrochloride removed by filtration. The dioxane was then removed by low pressure distillation at a temperature of 40-50°C and the viscous product remaining in the flask solved in absolute methyl alcohol (25 mL). After the vacuum distillation of the methyl alcohol, a sticky product resulted which turned into a solid product, after repeated washings with anhydrous ethyl ether. It was finally purified by recrystallization from boiling anhydrous methyl alcohol.
α-[N-di-(β-Chloroethyl)amide] of N’-(meta-formylaminobenzoyl)-D,L-asparagic acid (16). Light-yellow solid; yield 51.4 % (39 mg); m.p. 193-194°C. Anal. Calc. for C16H19Cl2N3O5: 47.54 % C, 4.74 % H, 10.39 % N, 17.54 % Cl; Found: 47.22 % C, 4.50 % H, 10.24 % N, 17.68 % Cl; IR (ν, cm-1): 3350, 3600 (NH); 1640, 1680 (CO); 770 (C-Cl); 705 (CH); 1H-NMR δ (CD3COCD3, 300 MHz): 2.60 (d, 2H, CH2); 4.20 (t, 2H, CH2); 4.80 (t, 1H, CH); 5.10 (t, 2H, CH2); 7.70 (t, 1H, Ar); 7.85 (d, 1H, Ar); 7.95 (d, 1H, Ar); 8.10 (s, 1H, Ar).
α-[N-di-(β-Chloroethyl)diamide] of N’-(meta-formylaminobenzoyl)-D,L-asparagic acid (17). Yellow solid; yield 50.5 % (40.5 mg); m.p. 198-200°C. Anal. calc. for C16H20Cl2N4O4: 47.65 % C, 5.00 % H, 13.89 % N, 17.58 % Cl; Found: 47.85 % C, 5.15 % H, 14.11 % N, 17.92 % Cl; IR (ν, cm-1): 3090, 3240 (NH); 1530, 1620 (CO); 745 (C-Cl); 710 (CH); 1H-NMR δ (CD3CN, 300 MHz): 2.90 (d, 2H, CH2); 4.40 (t, 2H, CH2); 4.95 (t, 1H, CH); 5.35 (t, 2H, CH2); 6.60 (s, 2H, NH2); 7.60 (t, 1H, Ar); 7.80 (d, 2H, Ar); 8.20 (s, 1H, Ar).
α-[N-di-(β-Chloroethyl)amide] of N’-(meta-formylaminobenzoyl)-D,L-methionine (18). Light brown solid; yield 56.38 % (47.30 mg); m.p. 177-178°C. Anal. calc. for C17H23Cl2N3O3S: 48.57 % C, 5.47 % H, 10.00 % N, 16.90 % Cl, 7.61 % S; Found: 48.31 % C, 5.40 % H, 9.85 % N, 16.83 % Cl, 7.41 % S; IR (ν, cm-1): 3065, 3472 (NH); 1700 (CO); 771 (C-S); 801 (C-Cl); 848 (CH); 1H-NMR δ (DMSO-d6, 400 MHz): 2.10 (s, 3H, CH3); 2.56 (m, 4H, CH2); 3.50 (t, 4H, CH2); 3.82 (t, 4H, CH2); 4.56 (t, 1H, CH); 6.20 (d, 1H, NH); 7.80 (t, 1H, Ar); 8.43 (d, 1H, Ar); 8.48 (d, 1H, Ar); 8.73 (s, 1H, Ar); 9.07 (d, 1H, NH); 9.68 (d, 1H, CH).
α-[N-di-(β-Chloroethyl)amide] of N’-(meta-acetylaminobenzoyl)-D,L-asparagic acid (19). Grey solid; yield 52.8 % (44 mg); m.p. 98-100°C. Anal. calc. for C17H21Cl2N3O5: 48.82 % C, 5.06 % H, 10.05 % N, 16.95 % Cl; Found: 48.35 % C, 5.32 % H, 10.23 % N, 17.12 % Cl; IR (ν, cm-1): 3340, 3500 (NH); 1640, 1660 (CO); 750 (C-Cl); 698 (CH); 1H-NMR δ (CD3CN, 300 MHz): 2.90-3.00 (m, 2H, CH2); 4.95 (t, 1H, CH); 5.80 (s, 1H, NH); 7.65 (t, 1H, Ar); 7.95 (d, 2H, Ar); 8.20 (s, 1H, Ar).
α-[N-di-(β-Chloroethyl)diamide] of N’-(meta-acetylaminobenzoyl)-D,L-asparagic acid (20). Yellow solid; yield 53.8 % (44.8 mg); m.p. 168-169°C. Anal. calc. for C17H22Cl2N4O4: 48.93 % C, 5.31 % H, 13.43 % N, 16.99 % Cl; Found: 49.10 % C, 5.88 % H, 13.70 % N, 17.40 % Cl; IR (ν, cm-1): 3125, 3300 (NH); 1535, 1630 (CO); 740 (C-Cl); 698 (CH); 1H-NMR δ (CD3CN, 300 MHz): 2.75 (d, 2H, CH2); 4.50 (t, 2H, CH2); 4.90 (t, 1H, CH); 5.30 (t, 2H, CH2); 6.60 (s, 2H, NH2); 7.70 (t, 1H, Ar); 7.90 (d, 2H, Ar); 8.20 (s, 1H, Ar).
α-[N-di-(β-Chloroethyl)amide] of N’-(meta-acetylaminobenzoyl)-D,L-methionine (21). Light brown solid; yield 58.3 % (50.6 mg); m.p. 183-184°C. Anal. calc. for C18H25Cl2N3O3S: 49.76 % C, 5.76 % H, 9.67 % N, 16.35 % Cl, 7.37 % S; Found: 49.58 % C, 5.62 % H, 9.46 % N, 16.17 % Cl, 7.28 % S; IR (ν, cm-1): 3010, 3328 (NH); 1685 (CO); 718 (C-S); 670 (C-Cl); 855 (CH); 1H-NMR δ (DMSO-d6, 400 MHz): 2.11 (s, 3H, CH3); 2.30 (s, 3H, CH3); 2.56 (m, 4H, CH2); 3.28 (t, 4H, CH2); 3.85 (t, 4H, CH2); 4.56 (t, 1H, CH); 6.02 (s, 1H, NH); 7.80 (t, 1H, Ar); 8.32 (d, 1H, Ar); 8.42 (d, 1H, Ar); 8.73 (s, 1H, Ar); 9.08 (d, 1H, NH).
Toxicity study
The acute toxicity, the DL
50 values respectively, was estimated by intraperitoneal administration of the compounds as a suspension in 1% methylcellulose to groups of four mice each, weighting 20±2 g, according to the classical laboratory procedure [
21]. The methylcellulose was chosen in these experiments since it is chemically inert and inactive on the animal tissue [
22]. The tested animals were carefully followed and the death rate registered seven days later.
Antitumour activity
The antitumour activity was estimated by using two experimental tumors: Ehrlich ascite and Walker 256 carcinosarcoma. Mice A
2G, weighing 25-30 g (± 2g) and Wistar rats, weighing 100-130 g (± 15 g), were used. The Ehrlich ascite tumors were transplanted to mice and the Walker 256 carcinosarcoma to rats. The ascite was transplanted intraperitoneally, from donors with 14 day old while the solid tumors was transplanted subcutaneously from donors with 21 day old tumors. The substances were administrated intraperitoneally, as suspensions in 1% methylcellulose. Single injection was made seven days after the transplantation with ascite and fourteen days after the transplantation with solid tumour. Concentrations of 400 mg, 200 mg and 40 mg/ kg body were taken and groups of 20 animals used with every concentration. The reference group of 20 animals with tumours was taken for every tumoural series. The inhibition induced by the compounds under study was estimated according to the literature method [
9,
11,
23] after seven and fourteen days from their administration for the ascite and for solid tumour respectively.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
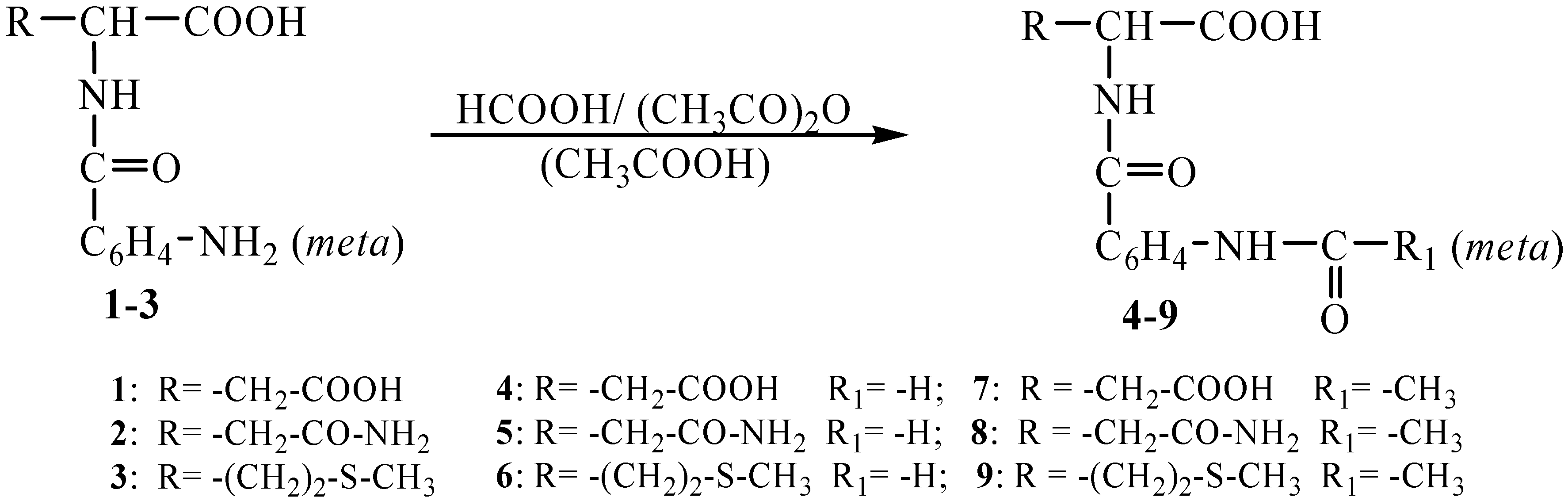{kind=link}
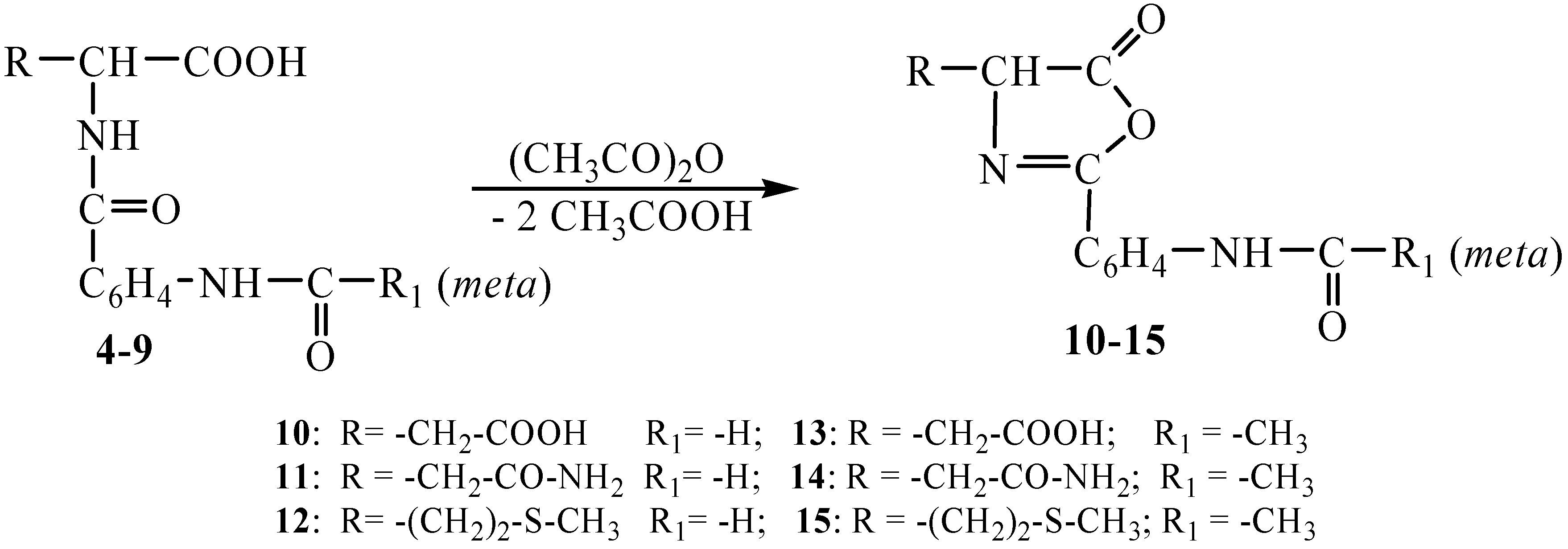{kind=link}
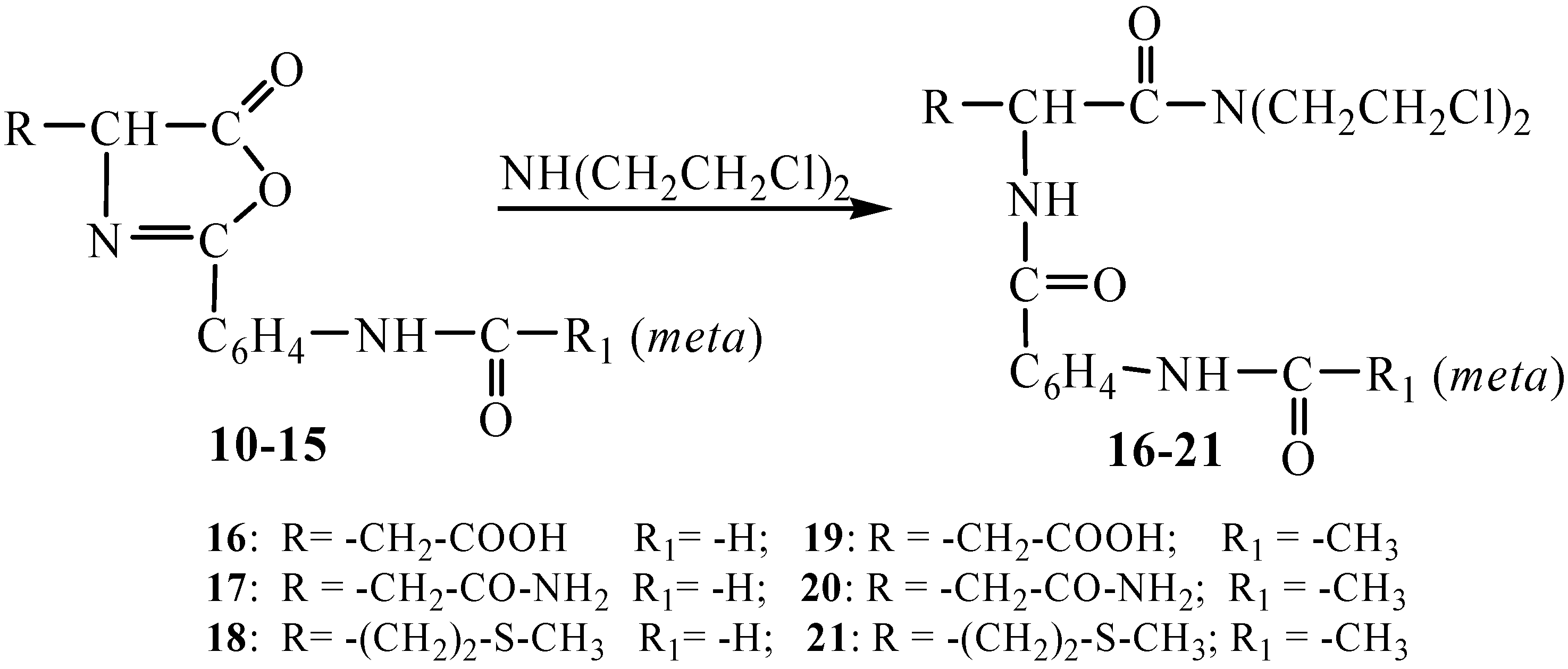{kind=link}
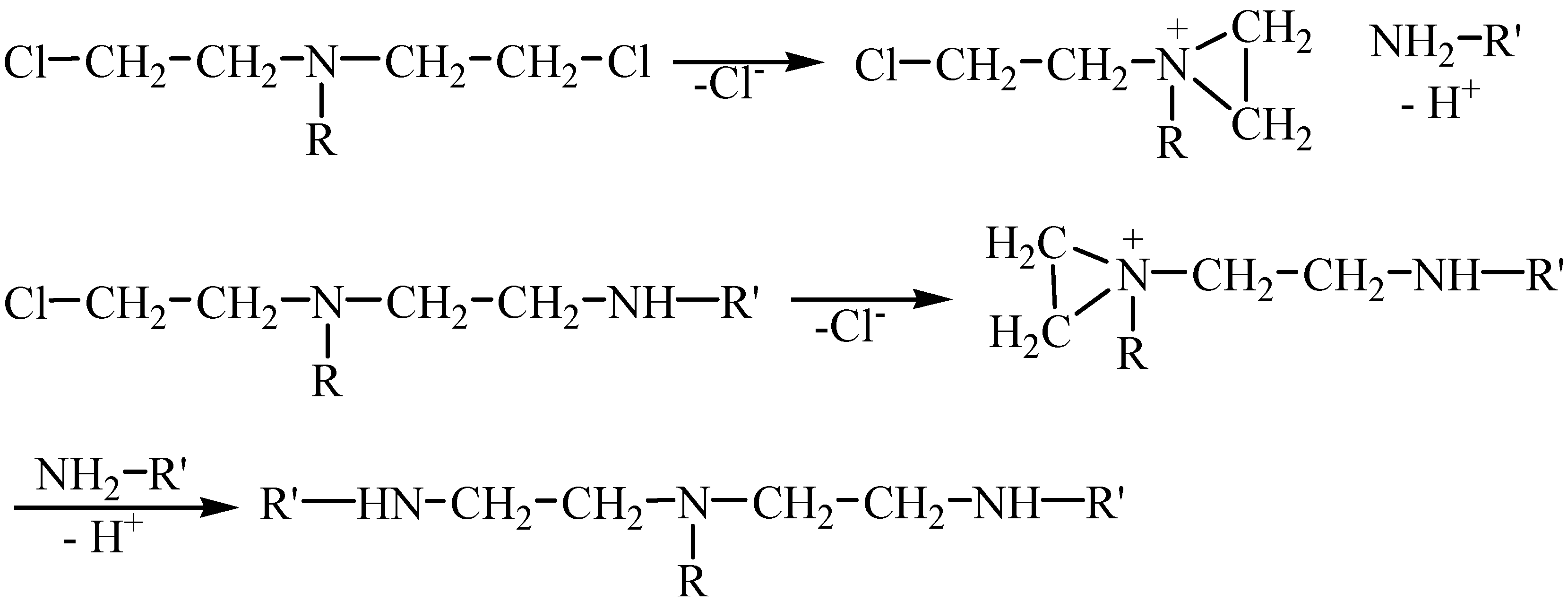{kind=link}
