Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. 5-Fluorouracil delivery systems
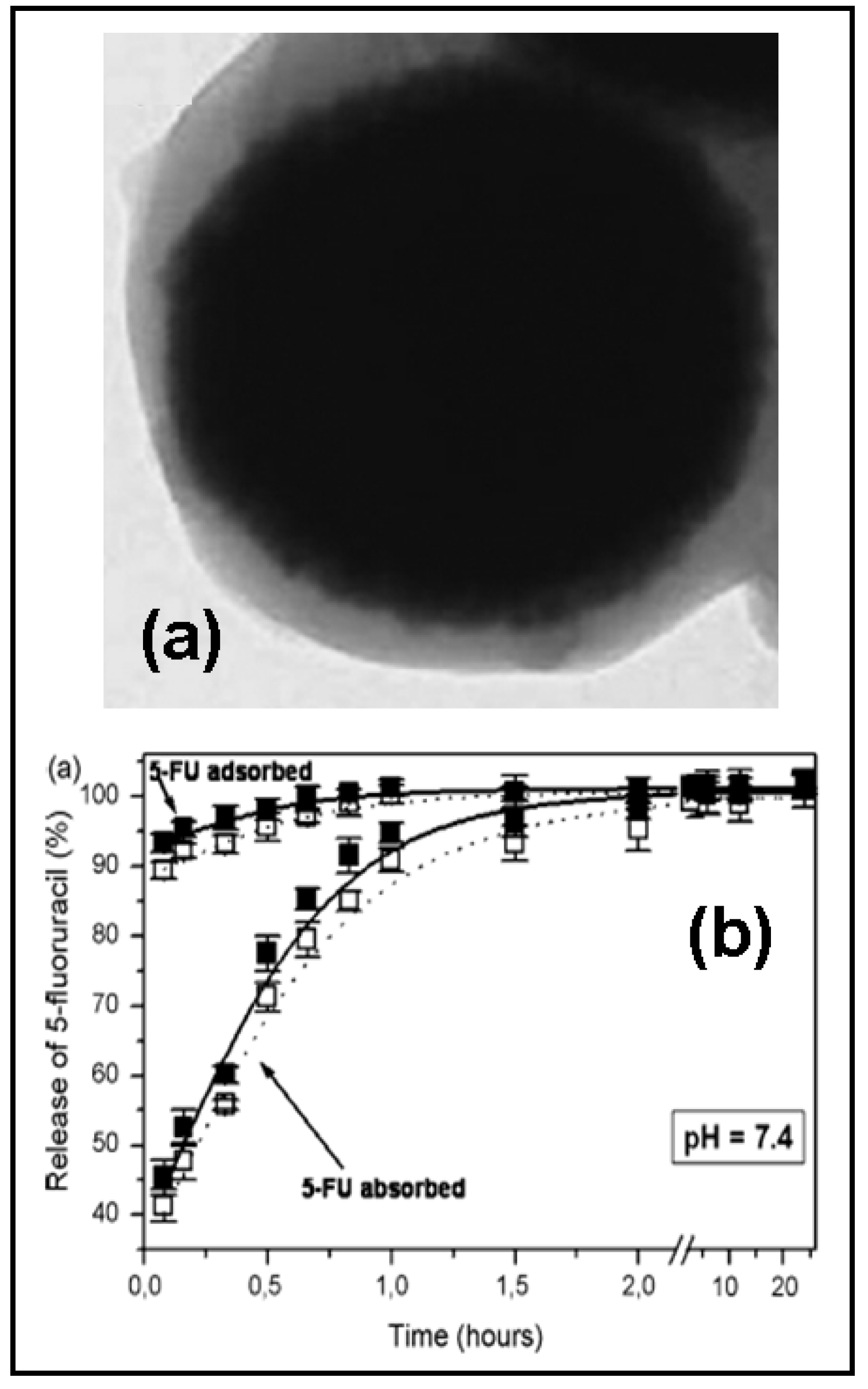
2.1 Biodegradable polymeric particles
2.1.1 Alginate beads
2.1.2 Poly(ε-caprolactone)
2.1.3. Chitosan
2.1.4. Eudragit®
2.1.5. Guar gum
2.1.6. Gelatin
2.1.7. Poly(alkylcyanoacrylates)
2.1.8. Poly(glutaraldehyde)
2.1.9. Polymers based on methacrylic acid
2.1.10. Poly(α-malic acid)
2.1.11. Poly(methilidene malonate 2.1.2)
2.1.12. Polyacrylamide
2.1.13. Poly(ortho-ester)s (POE)
2.1.14. Poly(D,L-lactide) (PLA) and poly(D,L-lactide-co-glycolide) (PLGA)
2.1.15. Dendrimers
2.2. Hydrogels
2.3. Vesicular systems: liposomes and niosomes
2.3.1. Liposomes
2.3.2. Niosomes
2.4. Magnetic drug delivery systems
2.5. Lipoproteins
2.6. Clay minerals and anionic clays
2.7. Metals
2.8. Ion exchange resins
3. Preclinical studies with 5-fluorouracil delivery systems
4. Conclusions
References
- Wong, H.L.; Bendayan, R.; Rauth, A.M.; Li, Y.; Wu, X.Y. Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Advan. Drug Delivery Rev. 2007, 59, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Durán, J.D.G.; Arias, J.L.; Gallardo, V.; Delgado, A.V. Magnetic colloids as drug vehicles. J. Pharm. Sci. 2008, 97, 2948–2983. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, Y.; Xu, S.-J.; Chen, W.-S. 5-fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Ruiz, M.A.; López-Viota, M.; Delgado, A.V. Poly(alkylcyanoacrylate) colloidal particles as vehicles for antitumour drug delivery: A comparative study. Colloids Surf. B Biointerfaces 2008, 62, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Galllardo, V.; Gómez-Lopera, S.A.; Plaza, R.C.; Delgado, A.V. Synthesis and characterization of poly(ethyl-2-cyanoacrylate) nanoparticles with a magnetic core. J. Control. Release 2001, 77, 309–321. [Google Scholar] [CrossRef]
- Arica, B.; Çaliş, S.; Kaş, H.S.; Sargon, M.F.; Hincal, A.A. 5-fluorouracil encapsulated alginate beads for the treatment of breast cancer. Int. J. Pharm. 2002, 242, 267–269. [Google Scholar] [CrossRef]
- Martini, L.G.; Collett, J.H.; Attwood, D. The release of 5-fluorouracil from microspheres of poly(epsilon-caprolactone-co-ethylene oxide). Drug Dev. Ind. Pharm. 2000, 26, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.D.; Cerrai, P.; Tricoli, M.; Maltinti, S. Release of 5-fluorouracil by biodegradable poly(ester-ether-ester)s. Part I: release by fused thin sheets. J. Mater. Sci. Mater. Med. 2001, 12, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Martini, L.G.A.; Collett, J.H.; Attwood, D. The release of 5-fluorouracil from a swellable matrix of a tri block copolymer of ε-caprolactone and ethylene oxide. Pharm. Res. 1995, 12, 1786–1790. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Chung, D.; Jeongho, A. Poly(D,L-lactide-ran-ε-caprolactone)-poly(ethylene glycol)-poly(D,L-lactide-ran-ε-caprolactone) as parenteral drug-delivery systems. Biomaterials 2004, 25, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Niu, G.C.-C.; Kuo, S.M.; Chen, S.F. Preparation and preliminary characterization of concentric multi-walled chitosan microspheres. J. Biomed. Mat. Res. A 2006, 81, 554–566. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.R.; Parikh, R.H. Two-stage optimization process for formulation of chitosan microspheres. AAPS Pharm. Sci. Tech. 2004, 5, E5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-Y.; Shen, X.-Z.; Wang, J.-Y.; Dong, L.; Zheng, Y.-L.; Wu, L.-L. Preparation of chitosan-polyaspartic acid-5-fluorouracil nanoparticles and its anti-carcinoma effect on tumor growth in nude mice. World J. Gastroenterol. 2008, 14, 3554–3562. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Chen, D.; Gu, J.; Qin, J. Nanoparticles of 5-fluorouracil (5-FU) loaded N-succinyl (Suc-Chi) for cancer chemotherapy: preparation, characterization – in vivo drug release and antitumour activity. J. Pharm. Pharmacol. 2006, 58, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, W.; Wang, C.; Hu, J.; Fu, S.; Dong, L.; Wu, L.; Shen, X. Nanoparticles based on the complex of chitosan and polyaspartic acid sodium salt: preparation, characterization and the use for 5-fluorouracil delivery. Eur. J. Pharm. Biopharm. 2007, 67, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Rokhade, A.P.; Shelke, N.B.; Patil, S.A.; Aminabhavi, T.M. Novel hydrogel microspheres of chitosan and pluronic F-127 for controlled release of 5-fluorouracil. J. Microencapsul. 2007, 24, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, Q.; Luo, H. Preparation and characterization of N-(2-carboxybenzyl)chitosan as a potential pH-sensitive hydrogel for drug delivery. Carbohydr. Res. 2007, 342, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Chandy, T.; Das, G.S.; Rao, G.H. 5-fluorouracil-loaded chitosan coated polylactic acid microspheres as biodegradable drug carriers for cerebral tumours. J. Microencapsul. 2000, 17, 625–638. [Google Scholar] [PubMed]
- Zambito, Y.; Baggiani, A.; Carelli, V.; Serafín, M.F.; Di Colo, G. Matrices for site-specific controlled-delivery of 5-fluorouracil to descending colon. J. Control. Release 2005, 102, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, A.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Microsphere design for the colonic delivery of 5-fluorouracil. J. Control. Release 2003, 90, 313–322. [Google Scholar] [CrossRef]
- Lamprecht, A.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Observations in simultaneous microencapsulation of 5-fluorouracil and leucovorin for combined pH-dependent release. Eur. J. Pharm. Biopharm. 2005, 59, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Krishnaiah, Y.S.R.; Satyanarayana, V.; Kumar, B.D.; Karthikeyan, R.S. In vitro drug release studies on guar gum-based colon targeted oral drug delivery systems of 5-fluorouracil. Eur. J. Pharm. Sci. 2002, 16, 185–192. [Google Scholar] [CrossRef]
- Harivardhan Reddy, L. Handbook of Particulate Drug Delivery; Ravi Kumar, M.N.V., Ed.; American Scientific Publishers: California, USA, 2008; Vol. 2, Chapter 3; p. 41. [Google Scholar]
- Narayani, R.; Rao, P. Gelatin microsphere cocktails of different sizes for the controlled release of anticancer drugs. Int. J. Pharm. 1996, 143, 255–258. [Google Scholar] [CrossRef]
- Couvreur, P.; Kante, B.; Roland, M.; Guiot, P.; Bauduin, P.; Speiser, P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: preparation, morphological and sorptive properties. J. Pharm. Pharmacol. 1979, 31, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, M.; Velichkova, R.; Ivanova, G.; Enchev, V.; Abrahams, I. Poly(butylcyanoacrylate) nanoparticles for topical delivery of 5-fluorouracil. Int. J. Pharm. 2003, 263, 133–140. [Google Scholar] [CrossRef]
- Simeonova, M.; Velichkova, R.; Ivanova, G.; Enchev, V.; Abrahams, I. Study on the role of 5-fluorouracil in the polymerization of butylcyanoacrylate during the formation of nanoparticles. J. Drug Target 2004, 12, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J.; Hartmann, H.R. Comparative study on the cytostatic effects and the tissue distribution of 5-fluorouracil in a free form and bound to polybutylcyanoacrylate nanoparticles in sarcoma 180-bearing mice. Oncology 1983, 40, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Hadjikirova, M.; Troyanova, P.; Simeonova, M. Nanoparticles as drug carrier system of 5-fluorouracil in local treatment of patients with superficial basal cell carcinoma. J. BUON 2005, 10, 517–521. [Google Scholar] [PubMed]
- Mukherji, G.; Murthy, R.S.R.; Miglani, B.D. Preparation and evaluation of polyglutaraldehyde nanoparticles containing 5-fluorouracil. Int. J. Pharm. 1989, 50, 15–19. [Google Scholar] [CrossRef]
- Denizli, A.; Kiremitci, M.; Pişkin, E. Subcutaneous polymeric matrix system p(HEMA-BGA) for controlled release of an anticancer drug (5-fluorouracil). Biomaterials 1988, 9, 363–366. [Google Scholar] [CrossRef]
- Singh, B.; Chauhan, N. Preliminary evaluation of molecular imprinting of-5-fluorouracil within hydrogels for use as drug delivery systems. Acta Biomater. 2008, 4, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Puoci, F.; Iemma, F.; Cirillo, G.; Picci, N.; Matricardi, P.; Alhaique, F. Molecularly imprinted polymers for 5-fluorouracil release in biological fluids. Molecules 2007, 12, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Ohya, Y.; Kobayashi, H.; Ouchi, T. Design of poly(α-malic acid)-5FU-saccharide conjugate exhibiting antitumor activity. React. Polym. 1991, 15, 153–163. [Google Scholar] [CrossRef]
- Ouchi, Y.; Fujino, A.; Tanaka, K.; Banba, T. Synthesis and antitumor activity of conjugates of poly(α-malic acid) and 5-fluorouracil bound via ester, amide or carbamoyl bonds. J. Control. Release 1990, 12, 143–153. [Google Scholar] [CrossRef]
- Fournier, E.; Passirani, C.; Vonarbourg, A.; Lemaire, L.; Colin, N.; Sagodira, S.; Menei, P.; Benoit, J.-P. Therapeutic efficacy study of novel 5-FU-loaded PMM 2.1.2-based microspheres on C6 glioma. Int. J. Pharm. 2003, 268, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Fournier, E.; Passirani, C.; Colin, N.; Breton, P.; Sagodira, S.; Benoit, J.-P. Development of novel 5-FU-loaded poly(methylidene malonate 2.1.2)-based microspheres for the treatment of brain cancers. Eur. J. Pharm. Biopharm. 2004, 57, 189–197. [Google Scholar] [CrossRef]
- Sairam, M.; Babu, V.R.; Naidu, B.V.K.; Aminabhavi, T.M. Encapsulation efficiency and controlled release characteristics of crosslinked polyacrylamide particles. Int. J. Pharm. 2006, 320, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.R.; Sairam, M.; Hosamani, K.M.; Aminabhavi, T.M. Development of 5-fluorouracil loaded poly(acrylamide-co-methylmethacrylate) novel core-shell microspheres: in vitro release studies. Int. J. Pharm. 2006, 325, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Zignani, M.; Einmahl, S.; Baeyens, V.; Varesio, E.; Veuthey, J.L.; Anderson, J.; Heller, J.; Tabatabay, C.; Gurny, R. A poly(ortho ester) designed for combine ocular delivery of dexamethasone sodium phosphate and 5-fluorouracil: subconjunctival tolerance and in vitro release. Eur. J. Pharm. Biopharm. 2000, 50, 251–255. [Google Scholar] [CrossRef]
- Einmahl, S.; Zignani, M.; Varesio, E.; Heller, J.; Veuthey, J.L.; Tabatabay, C.; Gurny, R. Concomitant and controlled release of dexamethasone and 5-fluorouracil from poly(ortho ester). Int. J. Pharm. 1999, 185, 189–198. [Google Scholar] [CrossRef]
- Sintzel, M.B.; Merkli, A.; Heller, J.; Tabatabay, C.; Gurny, R. Synthesis and analysis of viscous poly(ortho-ester) analogs for controlled drug release. Int. J. Pharm. 1997, 155, 263–269. [Google Scholar] [CrossRef]
- Ng, S.Y.; Shen, H.R.; Lopez, E.; Zherebin, Y.; Barr, J.; Schacht, E.; Heller, J. Development of a poly(ortho ester) prototype with a latent acid in the polymer backbone for 5-fluorouracil delivery. J. Control. Release 2000, 65, 367–374. [Google Scholar] [CrossRef]
- Bouassida, W.; Doyle, A.; Cohen, F.B.; Lteif, Y.; Labbe, A.; Lachkar, Y. Résultats à moyen terme de la chirurgie du glaucome avec polymère biodégradable libérant du 5-FU (in French). J. Fr. d´Ophtalmologie 2007, 30, 2S263. [Google Scholar]
- Lo, C.-L.; Lin, K.-M.; Hsiue, G.-h. Preparation and characterization of intelligent core-shell nanoparticles based on poly(D,L-lactide)-g-poly(N-isopropylacrylamide-co-methacrylic acid). J. Control. Release 2005, 104, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, S.S.; Jie, P.; Min, F.; Freddy, B.Y.C.; Leong-Huat, G. Micelle-like nanoparticles of PLA-PEG-PLA triblock copolymer as chemotherapeutic carrier. Int. J. Pharm. 2005, 298, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Jie, P.; Venkatraman, S.S.; Min, F.; Freddy, B.Y.C.; Huat, G.L. Micelle-like nanoparticles of star-branched PEO-PLA copolymers as chemotherapeutic carrier. J. Control. Release 2005, 110, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.J.; Zhang, J.X.; Wang, C.; Yasuda, H.; Ichimaru, A.; Yamamoto, K. Preparation and in vitro release of 5-fluorouracil-loaded microspheres based on poly(L-lactide) and its carbonate copolymers. J. Microencapsul. 2003, 20, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Parikh, R.H.; Parikh, J.R.; Dubey, R.R.; Soni, H.N.; Kapadia, K.N. Poly(D,L-lactide-coglycolide) microspheres containing 5-fluorouracil: optimization of process parameters. AAPS Pharm. Sci. Tech. 2003, 4, E13. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.D.; Sastre, R.L.; Teijón, C.; Olmo, R.; Teijón, J.M. 5-Fluorouracil-loaded microspheres prepared by spray-drying poly(D,L-lactide) and poly(lactide-co-glycolide) polymers: characterization and drug release. J. Microencapsul. 2005, 22, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Sastre, R.L.; Olmo, R.; Teijón, C.; Muñíz, E.; Teijón, J.M.; Blanco, M.D. 5-Fluorouracil plasma levels and biodegradation of subcutaneously injected drug-loaded microspheres prepared by spray-drying poly(D,L-lactide) and poly(D,L-lactide-co-glycolide) polymers. Int. J. Pharm. 2007, 338, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Bozkir, A.; Saka, O.M. Formulation and investigation of 5-FU nanoparticles with factorial design-based studies. Il Farmaco 2005, 60, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Takeuchi, H.; Hino, T.; Kunou, N.; Kawashima, Y. Preparations of biodegradable nanospheres of water-soluble and insoluble drugs with D,L-lactide/glycolide copolymer by a novel spontaneous emulsification solvent diffusion method, and the drug release behaviour. J. Control. Release 1993, 25, 89–98. [Google Scholar] [CrossRef]
- McCarron, P.A.; Woolfson, A.D.; Keating, S.M. Sustained release of 5-fluorouracil from polymeric nanoparticles. J. Pharm. Pharmacol. 2000, 52, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chae, G.S.; Kim, M.S.; Cho, S.H.; Lee, H.B.; Khang, G. Degradation behaviour in vitro for poly(D,L-lactide-co-glycolide) as drug carrier. Biomed. Mater. Eng. 2004, 14, 185–192. [Google Scholar] [PubMed]
- Hitzman, C.J.; Wiedmann, T.S.; Dai, H.; Elmquist, W.F. Measurement of drug release from microcarriers by microdialysis. J. Pharm. Sci. 2005, 94, 1456–1466. [Google Scholar] [CrossRef] [PubMed]
- Faisant, N.; Akiki, J.; Siepmann, F.; Benoit, J.P.; Siepmann, J. Effects of the type of release medium on drug release from PLGA-based microparticles: experiment and theory. Int. J. Pharm. 2006, 314, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Faisant, N.; Akiki, J.; Richard, J.; Benoit, J.P. Effect of the size of the biodegradable microparticles on drug release : experiment and theory. J. Control. Release 2004, 96, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Faisant, N.; Siepmann, J.; Oury, P.; Laffineur, V.; Bruna, E.; Haffner, J.; Benoit, J.-P. The effect of gamma-irradiation on drug release from bioerodible microparticles: a quantitative treatment. Int. J. Pharm. 2002, 242, 281–284. [Google Scholar] [CrossRef]
- Faisant, N.; Siepmann, J.; Richard, J.; Benoit, J.-P. Mathematical modeling of drug release from bioerodible microparticles: effect of gamma-irradiation. Eur. J. Pharm. Biopharm. 2003, 56, 271–279. [Google Scholar] [CrossRef]
- Boisdron-Celle, M.; Menei, P.; Benoit, J.-P. Preparation and characterization of 5-fluorouracilloaded microparticles as biodegradable anticancer drug carriers. J. Pharm. Pharmacol. 1995, 47, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Menei, P.; Boisdron-Celle, M.; Croué, A.; Guy, G.; Benoit, J.-P. Effect of stereotactic implantation of biodegradable 5-fluorouracil-loaded microspheres in healthy and C6 gliomabearing rats. Neurosurgery 1996, 39, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Menei, P.; Venier, M.C.; Gamelin, E.; Saint-André, J.; Hayek, G.; Jadaud, E.; Mercier, P.; Guy, G.; Benoit, J.-P. Local and sustained delivery of 5-fluorouracil from biodegradable microspheres for the radiosensitization of glioblastoma. Cancer 1999, 86, 325–330. [Google Scholar] [CrossRef]
- Dang, W.; Daviau, T.; Ying, P.; Zhao, Y.; Nowotnik, D.; Clow, C.; Tyler, B.; Brem, H. Effects of GLIADEL wafer initial molecular weight on the erosion of wafer and release of BCNU. J. Control. Release 1996, 42, 83–92. [Google Scholar] [CrossRef]
- Roullin, V.-G.; Deverre, J.-R.; Lemaire, L.; Hindré, F.; Vernier-Julienne, M.C.; Vienet, R.; Benoit, J.-P. Anti-cancer drug diffusion within living rat brain tissue: an experimental study using [3H](6)-5-fluorouracil-loaded PLGA microspheres. Eur. J. Pharm. Biopharm. 2002, 53, 293–299. [Google Scholar] [CrossRef]
- Menei, P.; Jadaud, E.; Faisant, N.; Boisdron-Celle, M.; Michalak, S.; Fournier, D.; Delhaye, M.; Benoit, J.-P. Stereotaxic implantation of 5-fluorouracil-releasing microspheres in malignant glioma. Cancer 2004, 100, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Benoit, J.-P.; Faisant, N.; Venier-Julienne, M.-C.; Menei, P. Development of microspheres for neurological disorders : from basics to clinical applications. J. Control. Release 2000, 65, 285–296. [Google Scholar] [CrossRef]
- Hagiwara, A.; Takahashi, T.; Sawai, K.; Sakakura, C.; Tsujimoto, H.; Imanishi, T.; Ohgaki, M.; Yamazaki, J.; Muranishi, S.; Yamamoto, A.; Fujita, T. Pharmacological effects of 5-fluorouracil microspheres on peritoneal carcinomatosis in animals. Br. J. Cancer 1996, 74, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Sakakura, C.; Tsujimoto, H.; Imanishi, T.; Ohgaki, M.; Yamasaki, J.; Sawai, K.; Takahashi, T.; Fujita, T.; Yamamoto, A.; Muranishi, S.; Ikada, Y. Selective delivery of 5-fluorouracil (5-FU) to i.p. tissues using 5-FU microspheres in rats. Anticancer drugs 1997, 8, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Beale, G.; Hughes, M.; Akhtar, S. Co-delivery of an antisense oligonucleotide and 5-fluorouracil using sustained release poly(lactide-co-glycolide) microsphere formulations for potential combination therapy in cancer. Int. J. Pharm. 2002, 234, 129–138. [Google Scholar] [CrossRef]
- Gupte, A.; Ciftci, K. Formulation and characterization of Paclitaxel, 5-FU and Paclitaxel + 5-FU microspheres. Int. J. Pharm. 2004, 276, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xu, Y.; Chen, G.; Wei, P.; Ping, Q. PLGA nanoparticles for the oral delivery of 5-Fluorouracil using high pressure homogenization-emulsification as the preparation method and in vitro/in vivo studies. Drug Dev. Ind. Pharm. 2008, 34, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.K.; Tung, S.M.; Lu, D.W.; Chen, J.L.; Chiang, C.H. Formulation factors for preparing ocular biodegradable delivery systems of 5-fluorouracil microparticles. J. Microencapsul. 2001, 18, 507–519. [Google Scholar] [PubMed]
- Chiang, C.H.; Tung, S.M.; Lu, D.W.; Yeh, M.K. In vitro and in vivo evaluation of an ocular delivery system of 5-fluorouracil microspheres. J. Ocul. Pharmacol. Ther. 2001, 17, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Hitzman, C.J.; Elmquist, W.F.; Wattenberg, L.W.; Wiedmann, T.S. Development of a respirable sustained release microcarrier for 5-Fluorouracil I: in vitro assessment of liposomes, microspheres, and lipid coated nanoparticles. J. Pharm. Sci. 2006, 95, 1114–1126. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, R.X.; Du, B.; Lu, Z.R. In vitro release of 5-fluorouracil with cyclic core dendritic polymer. J. Control. Release 1999, 57, 249–257. [Google Scholar] [CrossRef]
- Bhadra, D.; Bhadra, S.; Jain, S.; Jain, N.K. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int. J. Pharm. 2003, 257, 111–124. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, H.; Yang, X.; Yang, Y. Drug release behavior from in situ geletinized thermosensitive nanogel aqueous dispersions. Int. J. Pharm. 2008, 361, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, K.; Šubr, V.; Seymour, L.W.; Duncan, R. Novel biodegradable hydrogels prepared using the divinylic crosslinking agent N,O-dimethacryloylhydroxylamine. 1. Synthesis and characterisation of rates of gel degradation, and rate of release of model drugs, in vitro and in vivo. J. Control. Release 1993, 24, 181–190. [Google Scholar] [CrossRef]
- Ravichandran, P.; Shantha, K.L.; Rao, K.P. Preparation, swelling characteristics and evaluation of hydrogels for stomach specific drug delivery. Int. J. Pharm. 1997, 154, 89–94. [Google Scholar] [CrossRef]
- Jeyanthi, R.; Rao, P. Controlled release of anticancer drugs from collagen-poly(HEMA) hydrogel matrices. J. Control. Release 1990, 13, 91–98. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Zhuo, R.-X.; Cui, J.-Z.; Zhang, J.-T. A novel thermo-responsive drug delivery system with positive controlled release. Int. J. Pharm. 2002, 235, 43–50. [Google Scholar] [CrossRef]
- Woolfson, A.D.; McCafferty, D.F.; McCarron, P.A.; Price, J.H. A bioadhesive patch cervical drug delivery system for the administration of 5-fluorouracil to cervical tissue. J. Control. Release 1995, 35, 49–58. [Google Scholar] [CrossRef]
- Sasaki, H.; Matsukawa, Y.; Hashida, M.; Sezaki, H. Characterization of alkylcarbamoyl derivatives of 5-fluorouracil and their application to liposome. Int. J. Pharm. 1987, 36, 147–156. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, N.; Aiguo, L.; Zou, W.; Xu, W. Preparation and evaluation of N3-O-toluylloaded liposomes. Int. J. Pharm. 2008, 353, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Elorza, B.; Elorza, M.A.; Frutos, G.; Chantres, J.R. Characterization of 5-fluorouracil loaded liposomes prepared by reverse-phase evaporation or freezing-thawing extrusion methods: study of drug release. Biochim. Biophys. Acta 1993, 1153, 135–142. [Google Scholar] [CrossRef]
- Wang, T.; Deng, Y.; Geng, Y.; Gao, Z.; Zou, J.; Wang, Z. Preparation of submicron unilamellar liposomes by freeze-drying double emulsions. Biochim. Biophys. Acta 2006, 1758, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Nii, T.; Ishii, F. Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int. J. Pharm. 2005, 298, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Fresta, M.; Villari, A.; Puglisi, G.; Cavallaro, G. 5-Fluorouracil : various kinds of loaded liposomes : encapsulation efficiency, storage stability and fusogenic properties. Int. J. Pharm. 1993, 99, 145–156. [Google Scholar] [CrossRef]
- Ozer, A.Y.; Talsma, H. Preparation and stability of liposomes containing 5-fluorouracil. Int. J. Pharm. 1989, 55, 185–191. [Google Scholar] [CrossRef]
- Glavas-Dodov, M.; Fredro-Kumbaradzi, E.; Goracinova, K.; Simonoska, M.; Calis, S.; Trajkovic-Jolevska, S.; Hincal, A.A. The effects of lyophilization on the stability of liposomes containing 5-FU. Int. J. Pharm. 2005, 291, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Soni, V.; Kohli, D.V.; Jain, S.K. Transferrin coupled liposomes as drug delivery carriers for brain targeting of 5-florouracil. J. Drug Target. 2005, 13, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Muzzalupo, R.; Nicoletta, F.P.; Trombino, S.; Cassano, R.; Iemma, F.; Picci, N. A new crown ether as vesicular carrier for 5-fluorouracil: Synthesis, characterization and drug delivery evaluation. Colloids Surf. B Biointerfaces 2007, 58, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Cosco, D.; Muzzalupo, R.; Trapasso, E.; Picci, N.; Fresta, M. Innovative bolasurfactant niosomes as topical delivery systems of 5-fluorouracil for the treatment of skin cancer. Int. J. Pharm. 2008, 353, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Gallardo, V.; Gómez-Lopera, S.A.; Delgado, A.V. Loading of 5-fluorouracil to poly(ethyl-2-cyanoacrylate) nanoparticles with a magnetic core. J. Biomed. Nanotech. 2005, 1, 214–223. [Google Scholar] [CrossRef]
- Arias, J.L.; Linares-Molinero, F.; Gallardo, V.; Delgado, A.V. Study of carbonyl iron/poly(butylcyanoacrylate) (core/shell) particles as anticancer drug delivery systems. Loading and release properties. Eur. J. Pharm. Sci. 2008, 33, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Gallardo, V.; Ruiz, M.A.; Delgado, A.V. Magnetite/poly(alkylcyanoacrylate) (core/shell) nanoparticles as 5-Fluorouracil delivery systems for active targeting. Eur. J. Pharm. Biopharm. 2008, 69, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Viroonchatapan, E.; Sato, H.; Ueno, M.; Adachi, I.; Tazawa, K.; Horikoshi, I. Release of 5-fluorouracil from thermosensitive magnetoliposomes induced by an electromagnetic field. J. Control. Release 1997, 46, 263–271. [Google Scholar] [CrossRef]
- Nagaich, S.; Khopade, A.J.; Jain, N.K. Lipid grafts of egg-box complex: a new supramolecular biovector for 5-fluorouracil delivery. Pharm. Acta Helv. 1999, 73, 227–236. [Google Scholar] [CrossRef]
- Kader, A.; Pater, A. Loading anticancer drugs into HDL as well as LDL has little affect on properties of complexes and enhances cytotoxicity to human carcinoma cells. J. Control. Release 2002, 80, 29–44. [Google Scholar] [CrossRef]
- Lin, F.H.; Lee, Y.H.; Jian, C.H.; Wong, J.-M.; Shieh, M.-J.; Wang, C.-Y. A study of montmorillonite intercalated with 5-fluorouracil as drug carrier. Biomaterials 2002, 23, 1981–1987. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, E.; Gao, L.; Xu, L. Synthesis and properties of Mg2Al layered double hydroxides containing 5-fluorouracil. J. Solid State Chem. 2005, 178, 736–741. [Google Scholar] [CrossRef]
- Selvaraj, V.; Alagar, M. Analytical detection and biological assay of antileukemic drug 5-fluorouracil using gold nanoparticles as a probe. Int. J. Pharm. 2007, 337, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Burton, M.A.; Gray, B.N. In vitro release of cytotoxic agents from ion exchange resins. J. Control. Release 1989, 8, 251–257. [Google Scholar] [CrossRef]
- Sample Availability: Not available.



© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arias, J.L. Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems. Molecules 2008, 13, 2340-2369. https://doi.org/10.3390/molecules13102340
Arias JL. Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems. Molecules. 2008; 13(10):2340-2369. https://doi.org/10.3390/molecules13102340
Chicago/Turabian StyleArias, José L. 2008. "Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems" Molecules 13, no. 10: 2340-2369. https://doi.org/10.3390/molecules13102340
APA StyleArias, J. L. (2008). Novel Strategies to Improve the Anticancer Action of 5-Fluorouracil by Using Drug Delivery Systems. Molecules, 13(10), 2340-2369. https://doi.org/10.3390/molecules13102340