Experimental
General
Triphenylphosphine, dialkyl acetylenedicarboxylate and 2-acetyl cyclopentanone were obtained from Fluka (Buchs, Switzerland) and were used without further purification. Melting points were measured on an Electrotermal 9100 apparatus and are uncorrected. 1H- and 13C-NMR spectra were measured with a Bruker DRX-500 AVANCE spectrometer at 500 and 125.8 MHz, respectively. Elemental analyses were performed using a Heraeus CHN-O-Rapid analyzer. IR spectra were recorded on a Shimadzu IR-470 spectrometer.
General procedure for preparation of dialkyl 2-(1-acetyl-2-oxocyclopentyl)-3-[1,1,1-triphenyl-λ5-phosphanylidene]succinates (exemplified by 3a)
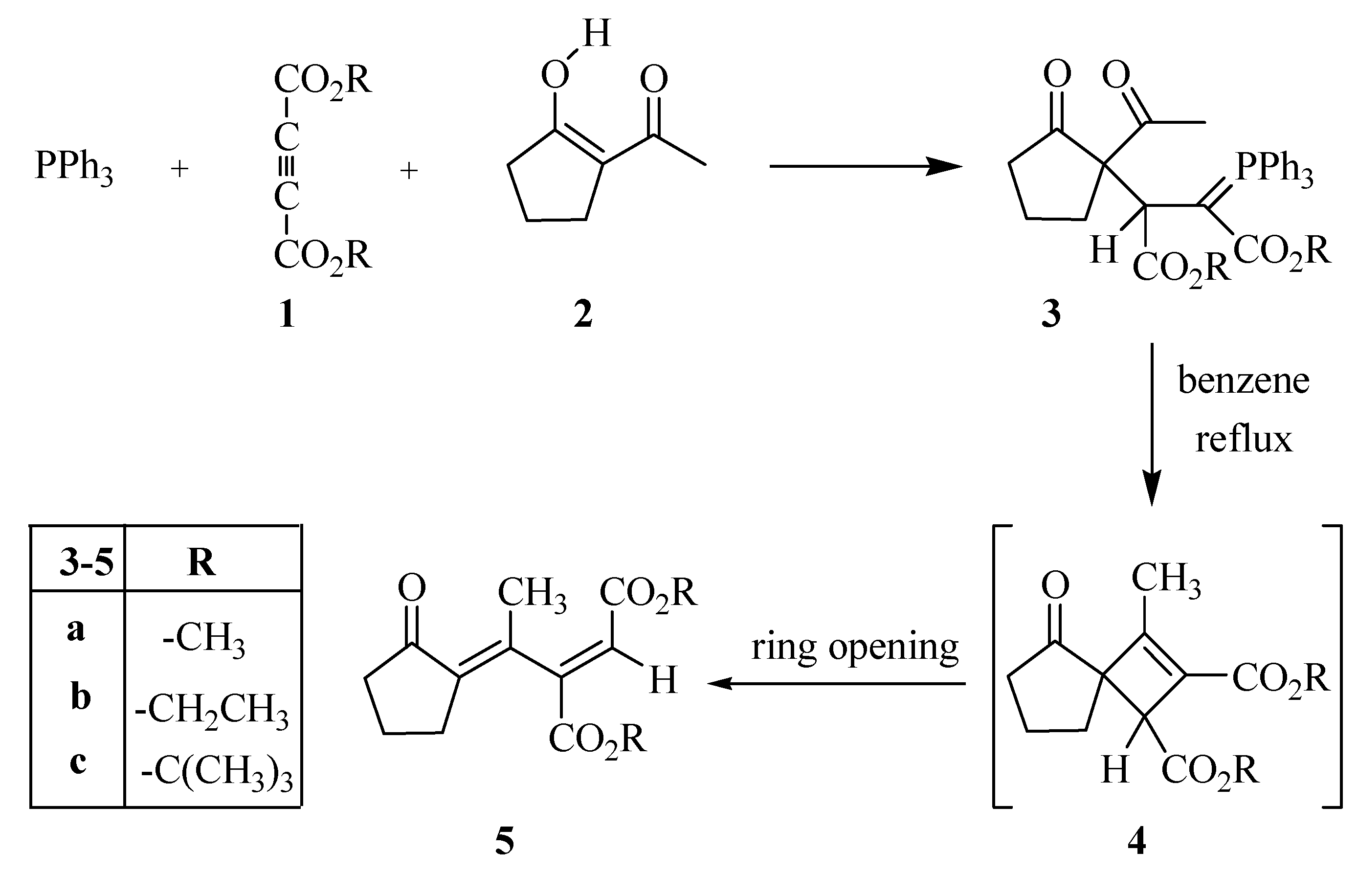
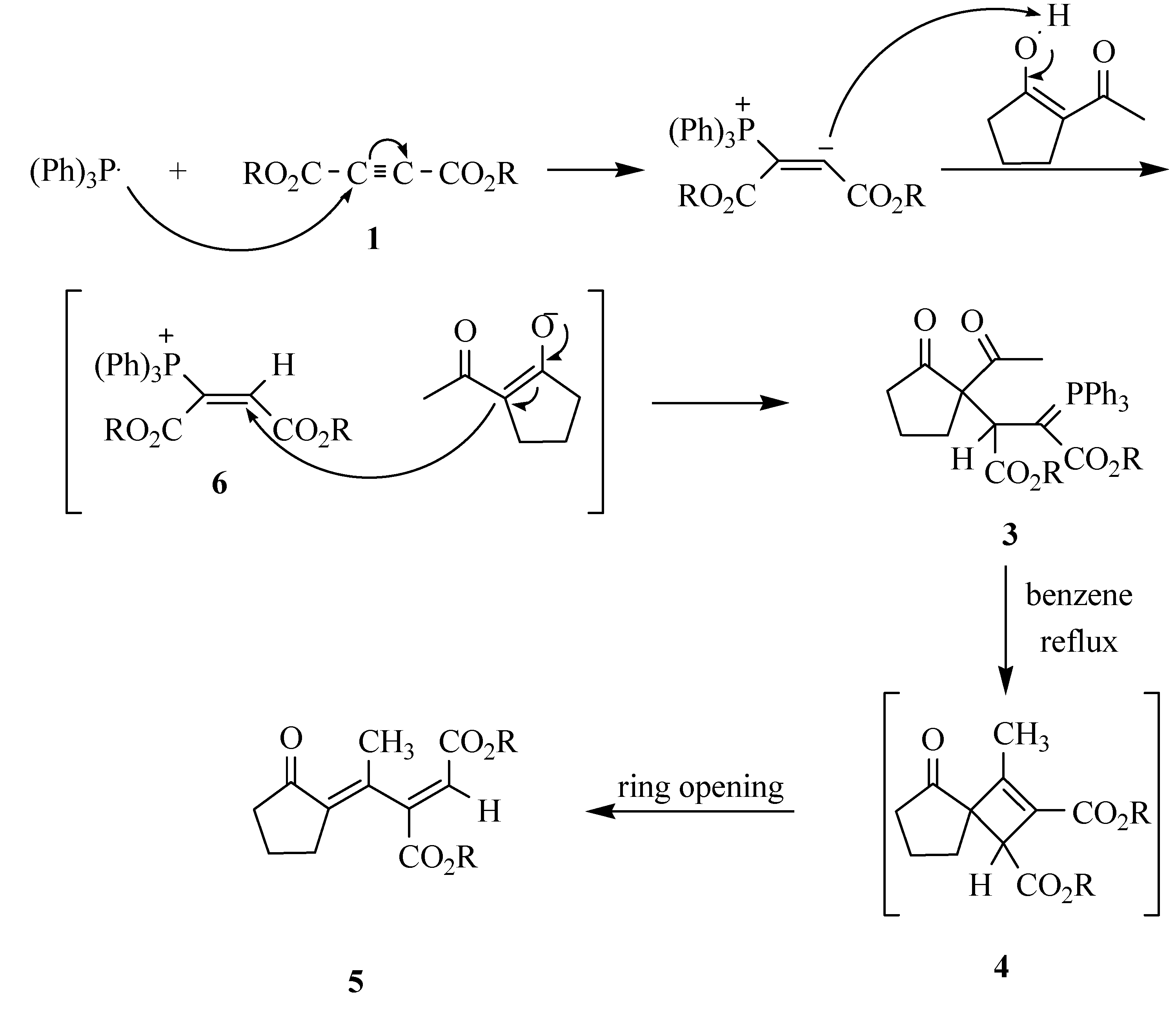
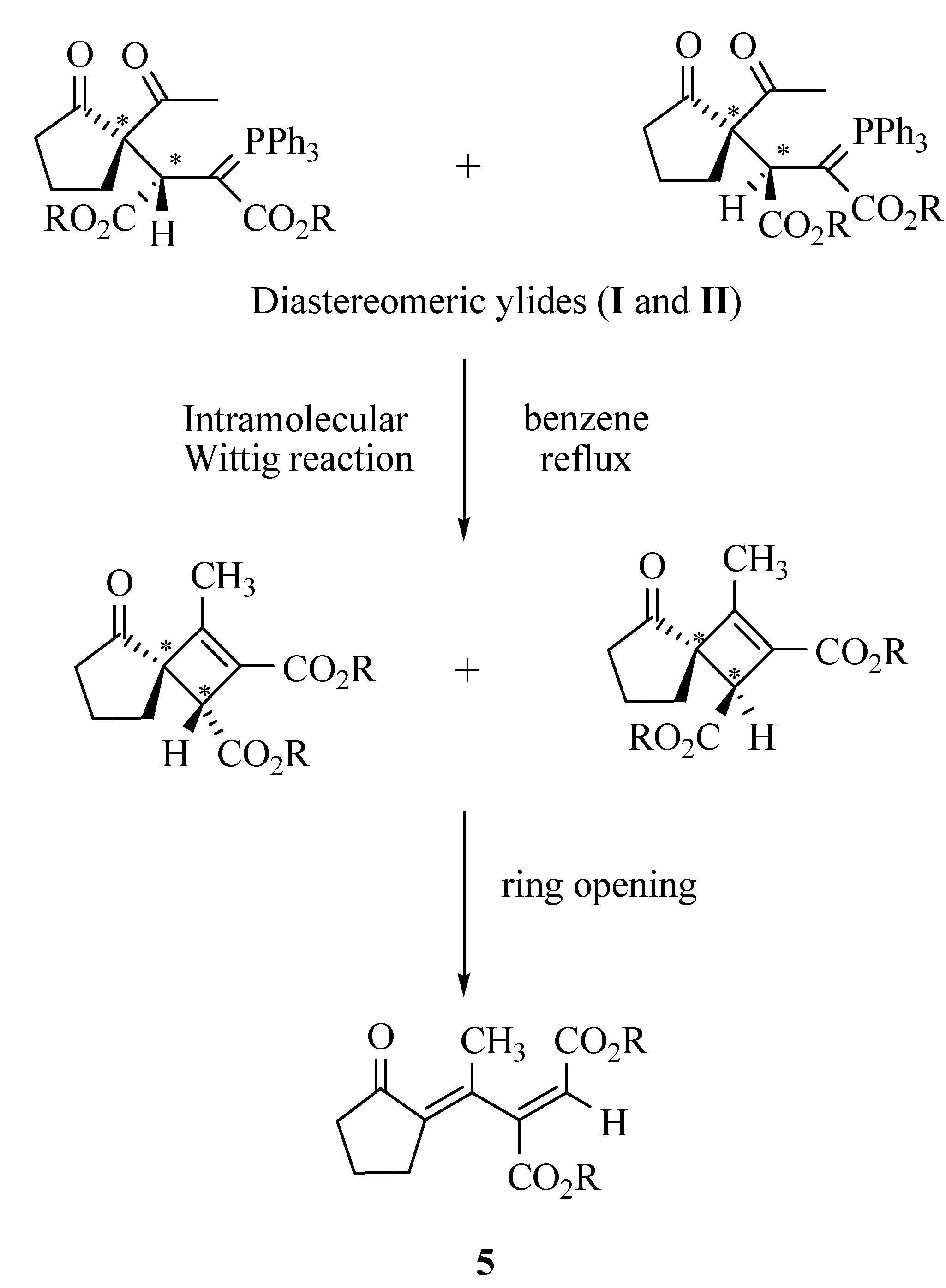
A mixture of dimethyl acetylenedicarboxylate (0.245 mL, 2 mmol) in CH2Cl2 (4 mL) was added dropwise at -10 ºC over 10 min. to a magnetically stirred solution of 2-acetylcyclopentanone (2, 0.252 g, 2 mmol) and triphenyl phosphine (0.524 g, 2 mmol) in CH2Cl2 (10 mL). The mixture was allowed to stand at room temperature along with stirring for 24 hours. The solvent was removed under reduced pressure and the residue was purified by silica gel column chromatography (Merck silica gel 60, 230-400 mesh) using ethyl acetate-hexane (30:70) as eluent. Two diastereomers were isolated. The solvents were removed under reduced pressure to give ylides 3a-I and 3a-II as white powders.
Dimethyl 2-(1-acetyl-2-oxocyclopentyl)-3-[1,1,1-triphenyl- λ5-phosphanylidene] succinate (3a)
First diastereomer (3a-I): M.p. 126-128.5 ºC; yield 50 %; IR (KBr, νmax, cm-1): 1755, 1730 and 1705 (C=O), 1629 (C=C); MS m/z (%): 530 (M+, 10), 499 (M+-OCH3, 14), 471 (M+-CO2Me, 25), 487 (M+-CH3CO, 18), 268 (M+- PPh3, 25), 209 [M+- (PPh3+CO2Me), 37], 43 (CH3CO+, 100); Anal. calcd. for C31H31O6P (530.56); C, 70.18; H, 5.89%. Found: C 70.06; H, 5.83%; 1H-NMR (CDCl3) 3a-I (Z): δH 1.42-1.50 (2H, m, CH2), 1.60 (3H, s, CH3), 1.97-2.13 (2H, m, CH2), 2.58-2.82 (2H, m, CH2), 2.92 and 3.70 (6H, 2s, 2OCH3), 3.56 (1H, d, 3JPH=18.4 Hz, CH), 7.4-7.7 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.12 (CH2), 27.64 (CH3), 29.81 (CH2), 37.31 (CH2), 39.38 (d, 1JPC = 122.5 Hz, P=C), 40.09 (d, 2JPC = 13.4 Hz, CH), 48.71 and 51.83 (2OCH3), 73.94 (cyclopentanone quaternary carbon), 127.61 (d, 1JPC = 91.4 Hz, Cipso), 128.58 (d, 3JPC = 11.7 Hz, Cmeta), 131.9 (Cpara), 133.97 (d, 2JPC = 9.4 Hz, Cortho), 169.94 (d, 2JPC = 13.1 Hz, C=O ester), 174.24 (d, 3JPC = 5.3 Hz, C=O ester), 203.70 and 216.22 (2C=O, ketones). 3a-I (E) (31 %) 1H-NMR (CDCl3): δH 1.55 (3H, s, CH3), 1.62-1.65 (2H, m, CH2), 2.2-2.40 (2H, m, CH2), 2.48-2.53 (2H, m, CH2), 3.43 and 3.68 (2OCH3), 3.59 (1H, d, 3JPH = 18.5 Hz, CH), 7.4-7.67 (15H, m, -Ph); 13C-NMR (CDCl3): δC 21.5 (CH2), 25.44 (CH3), 29.14 (CH2), 37.5 (d, 1JPC = 127.5 Hz, P=C), 38.70 (CH2), 48.12 (d, 2JPC = 13.2 Hz, CH), 49.65 and 52.34 (2OCH3), 73.26 (cyclo-pentanone quaternary carbon), 126.7 (d, 1JPC = 91.7 Hz, Cipso), 128.2 (d, 3JPC = 11.5 Hz, Cmeta), 131.6 (Cpara), 132.13 (d, 2JPC = 9.9 Hz, Cortho), 168.5 (d, 2JPC = 12.8 Hz, C=O ester), 174.8 (d, 3JPC = 5.2 Hz, C=O ester), 203.65 and 215.02 (2C=O, ketones).
Second diastereomer (3a-II): M.p. 151-152 ºC; yield 45 %; IR (KBr, νmax, cm-1): 3059 and 2987 (CH), 1751, 1726 and 1689 (C=O), 1629 (C=C); MS m/z (%): 530 (M+, 8), 471 (M+-CO2Me, 18), 487 (M+-CH3CO, 30), 268 (M+- PPh3, 21), 209 (M+- (PPh3+CO2Me), 28), 43 (CH3CO+, 100); Anal. calcd. for C31H31O6P (530.56); C, 70.18; H, 5.89%; Found C 70.05, H 5.78 %; 3a-II (Z) (68 %) 1H-NMR (CDCl3): δH 1.64 (3H, s, CH3), 1.82-1.95 (2H, m, CH2), 2.09-2.18 (2H, m, CH2), 2.88-2.96 (2H, m, CH2), 2.80 and 3.69 (6H, 2s, 2OCH3), 3.49 (1H, d, 3JPH = 18.8 Hz, CH), 7.45-7.64 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.33 (CH2), 25.45 (CH3), 29.12 (CH2), 39.84 (d, 1JPC = 121.9 Hz, CH), 40.07 (CH2), 48.37 (d, 2JPC = 13.4 Hz, CH), 51.82 and 52.22 (2OCH3), 73.97 (cyclopentanone quaternary carbon), 127.5 (d, 1JPC = 91.5 Hz, Cipso), 128.43 (d, 3JPC = 12.20 Hz, Cmeta), 131.92 (d, 4JPC = 2.9 Hz, Cpara), 134.02 (d, 2JPC = 9.5 Hz, Cortho), 170.39 (d, 2JPC = 13.3 Hz, C=O ester), 175 (d, 3JPC = 6.5 Hz, C=O ester), 202.8 and 215 (2C=O, ketones). 3a-II (E) (32 %) 1H-NMR (CDCl3): δH 1.65 (3H, s, CH3), 1.82-1.95 (2H, m, CH2), 2.09-2.18 (2H, m, CH2), 2.88-2.96 (2H, m, CH2), 3.47 and 3.69 (6H, 2s, 2OCH3), 3.64 (1H, d, 3JPH = 19.5 Hz, CH), 7.45-7.64 (15H, m, -Ph); 13C-NMR (CDCl3): δC 21.07 (CH2), 25.84 (CH3), 28.55 (CH2), 39.71 (CH2), 40.58 (d, 1JPC = 129.4 Hz, P=C), 48.37 (d, 2JPC = 13.4 Hz, CH), 51.82 and 52.22 (2OCH3), 73.97 (cyclopentanone quaternary carbon), 127.3 (d, 1JPC = 90.8 Hz, Cipso), 128.33 (d, 3JPC = 12.07 Hz, Cmeta), 131.97 (d, 4JPC = 2.83 Hz, Cpara), 132.12 (d, 2JPC = 9.3 Hz, Cortho), 171.07 (d, 2JPC = 13.5 Hz, C=O ester), 172.5 (d, 3JPC = 6.9 Hz, C=O ester), 202.3 and 214.8 (2C=O, ketones).
Diethyl 2-(1-acetyl-2-oxocyclopentyl)-3-[1,1,1-triphenyl- λ5-phosphanylidene] succinate (3b)
First diastereomer (3b-I): M.p. 157.5-159 ºC; yield 53 %; IR (KBr, νmax, cm-1): 1745, 1730 and 1705 (C=O), 1632 (C=C); MS m/z (%): 558 (M+, 7), 530 (M+-C2H5, 14), 515 (M+-CH3CO, 31), 485 (M+-CO2Et, 18), 296 (M+- PPh3, 20), 223 [M+- (PPh3+CO2Et), 37], 43 (CH3CO+, 100); Anal. calcd. for C33H35O6P (530.56); C, 70.96; H, 6.32%; Found: C 70.88; H, 6.25%; 3b-I (Z) (64 %) 1H-NMR (CDCl3): δH 1.18 (3H, t, 3JHH = 7 Hz, CH3), 1.25 (3H, t, 3JHH = 7.2 Hz, CH3), 1.51-1.55 (2H, m, CH2), 1.57 (3H, s, CH3), 1.85-1.97 (2H, m, CH2), 2.75-2.78 (2H, m, CH2), 3.56 (1H, d, 3JPH = 18.5 Hz, CH), 3.66-3.68 (2H, m, OCH2), 4.12-4.15 (2H, m, OCH2), 7.46-7.81 (15H, m, -Ph); 13C-NMR (CDCl3): δC 14.13 and 14.34 (2CH3), 20.17 and 27.70 (2CH2), 29.69 (CH3), 37.38 (d, 1JPC = 122.78 Hz, P=C), 37.78 (CH2), 49.12 (d, 2JPC = 12.96 Hz, CH), 57.54 and 61.35 (2OCH2), 74.17 (cyclopentanone quaternary carbon), 128.16 (d, 1JPC = 92.98 Hz, Cipso), 128.53 (d, 3JPC = 11.82 Hz, Cmeta), 131.86 (Cpara), 134.07 (d, 2JPC = 9.4 Hz, Cortho), 166.03 (d, 2JPC = 12.8 Hz, C=O ester), 173.87 (d, 3JPC = 6.3 Hz, C=O ester), 204.51 and 216.40 (2C=O, ketones). 3b-I (E) (36 %); 1H-NMR (CDCl3): δH 1.21 (3H, t, 3JHH = 7.1 Hz, CH3), 1.25 (3H, t, 3JHH = 7.2 Hz, CH3), 1.41-1.46 (2H, m, CH2), 1.62 (3H, s, CH3CO), 2.07-2.12 (2H, m, CH2), 2.44-2.52 (2H, m, CH2), 3.37-3.39 (2H, m, OCH2), 3.47 (1H, d, 3JPH = 19.8 Hz, CHCO2Et), 4.12-4.15 (2H, m, OCH2), 7.46-7.81 (15H, m, -Ph); 13C-NMR (CDCl3): δC 13.10 and 13.60 (2CH3), 19.28 and 25.86 (2CH2), 29.05 (CH3CO), 37.1 (d, 1JPC = 125.8 Hz, P=C), 37.26 (CH2CO), 48.5 (d, 2JPC = 12.6 Hz, CHCO2Et), 60.97 and 62.15 (2OCH2), 71.5 (cyclopentanone quaternary carbon), 127.8 (d, 1JPC = 90.8 Hz, Cipso), 129.89 (3JPC=12.4 Hz, Cmeta), 132.13 (2JPC=9.81 Hz, Cortho), 134.83 (Cpara), 165.04 (d, 2JPC=13.1 Hz, C=O ester), 170.72 (d, 3JPC = 6.7 Hz, C=O ester), 203.84 and 215.01 (2C=O, ketones).
Di-tert-butyl 2-(1-acetyl-2-oxocyclopentyl)-3-[1,1,1-triphenyl- λ5-phosphanylidene] succinate (3c)
First diastereomer (3c-I): M.p. 149-149.5 ºC; yield 53 %; IR (KBr, νmax, cm-1): 1752, 1733 and 1705 (C=O), 1620 (C=C); MS m/z (%): 614 (M+, 5), 571 (M+-CH3CO, 25), 558 (M+-C4H8, 16), 513 (M+-CO2tBu, 28), 456 [M+-(CO2tBu+C4H9), 38], 352 (M+- PPh3, 14), 251 [M+- (PPh3+CO2tBu), 37], 43 (CH3CO+, 100); Anal. calcd. for C37H43O6P (614.73): C 72.29, H 7.05 %; Found C 72.15, H 6.95 %; 3c-I (Z) (59 %) 1H-NMR (CDCl3): δH 0.84 (9H, s, CMe3), 1.47 (9H, s, CMe3), 1.49 (3H, s, CH3), 1.87-1.88 (2H, m, CH2), 2.01-2.06 (2H, m, CH2), 2.78-2.83 (2H, m, CH2), 3.37 (1H, d, 3JPH = 16.8 Hz, CH), 7.4-7.55 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.28 and 25.83 (2CH2), 28 and 28.44 (2CMe3), 28.21 (CH3), 37.58 (d, 1JPC = 120.79 Hz, P=C), 38.76 (CH2), 44.92 (d, 2JPC = 14.28 Hz, CH), 74.49 (cyclopentanone quaternary carbon), 80.65 and 81.67 (2CMe3), 125.8 (d, 1JPC = 85.6 Hz, Cipso), 128.52 (d, 3JPC = 12.1 Hz, Cmeta), 131.96 (Cpara), 132.1 (d, 2JPC = 9.94 Hz, Cortho), 169.47 (d, 3JPC = 12.8 Hz, C=O ester), 175.73 (d, 3JPC = 6.5 Hz, C=O ester), 204.19 and 216.98 (2C=O, ketones). 3c-I (E) (41 %) 1H-NMR (CDCl3): δH 1.36 (9H, s, CMe3), 1.40 (9H, s, CMe3), 1.54 (CH3CO), 2.01-2.06 (2H, m, CH2), 2.51-2.56 (2H, m, CH2), 2.90-2.94 (2H, m, CH2), 3.35 (1H, d, 3JPH = 16.8 Hz, CH), 7.65-7.78 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.28 and 25.16 (2CH2), 28 and 28.04 (2CMe3), 28.13 (CH3), 38.3 (d, 1JPC = 122 Hz, P=C), 38.29 (CH2), 49.1 (d, 2JPC = 14.1 Hz, CH), 74.11 (cyclopentanone quaternary carbon), 81.18 and 81.79 (2CMe3), 126.2 (d, 1JPC = 84.5 Hz, Cipso), 128.30 (d, 3JPC = 11.95 Hz, Cmeta), 131.66 (Cpara), 135.38 (d, 2JPC = 10.1 Hz, Cortho), 168.80 (d, 2JPC = 13 Hz, C=O ester), 172.56 (d, 3JPC = 6.5 Hz, C=O ester), 202.41 and 212.82 (2C=O, ketones).
Second diastereomer; (3c-II): M.p. 157.5-159ºC; yield 42 %; IR (KBr, νmax, cm-1): 1748, 1725 and 1695 (C=O), 1612 (C=C); Anal. calcd. for C37H43O6P (614.72): C 72.29, H 7.05 %; Found C 72.18, H 6.97 %. 3c-II (Z) (66 %) 1H-NMR (CDCl3): δH 0.85 and 1.50 (18H, 2s, 2CMe3), 1.56 (3H, s, CH3CO), 1.87-1.92 (2H, m, CH2), 1.97-2.03 (2H, m, CH2), 3.10-3.16 (2H, m, CH2), 3.31 (1H, d, 3JPH = 20 Hz, CH), 7.40-7.52 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.25 and 25.03 (2CH2), 28.26 and 28.31 (2CMe3), 28.80 (CH3CO), 38.47 (d, 1JPC = 122.3 Hz, P=C), 40.23 (CH2CO), 50.03 (d, 2JPC = 14.1 Hz, CH), 74.38 (d, 3JPC = 3.4 Hz, cyclopentanone quaternary carbon), 77.01 and 80.71 (2CMe3), 125.90 (d, 1JPC = 84.8 Hz, Cipso), 128.1 (d, 3JPC = 11.8 Hz, Cmeta), 131.95 (Cpara), 132.10 (d, 2JPC = 9.4 Hz, Cortho), 160.9 (d, 2JPC = 13.1 Hz, C=O ester), 173.66 (d, 3JPC = 6.8 Hz, C=O ester), 203.12 and 215.98 (2C=O, ketones). 3c-II (E) (34 %) 1H-NMR (CDCl3): δH 1.37 and 1.50 (18H, 2s, 2CMe3), 1.52 (3H, s, CH3CO), 2.05-2.19 (2H, m, CH2), 2.43-2.48 (2H, m, CH2), 2.89-2.98 (2H, m, CH2), 3.30 (1H, d, 3JPH = 21.9 Hz, CH), 7.82-8.1 (15H, m, -Ph); 13C-NMR (CDCl3): δC 20.40 and 24.88 (2CH2), 27.91 and 28.48 (2CMe3), 29.28 (CH3CO), 40.62 (d, 1JPC = 131.33 Hz, P=C), 39.42 (CH2CO), 49.39 (d, 2JPC = 13.8 Hz, CH), 74.02 (d, 3JPC = 3.5 Hz cyclopentanone quaternary carbon), 77.78 and 80.60 (2CMe3), 125.90 (d, 1JPC = 84.8 Hz, Cipso), 128.52 (d, 3JPC = 12.1 Hz, Cmeta), 131.79 (Cpara), 135.36 (d, 2JPC = 9.9 Hz, Cortho), 170.83 (d, 2JPC = 13.3 Hz, C=O ester), 173.42 (d, 3JPC = 7 Hz, C=O ester), 202.61 and 215.43 (2C=O, ketones).
Preparation of Dimethyl (E)-2-[1-(2-oxocyclopentylidene)ethyl]-2-butenedioate (5a)
Compound 3a (I or II) was refluxed in benzene for 24 hours. The solvent was removed under reduced pressure and the viscous residue was purified by silica gel column chromatography (Merck silica gel 60, 230-400 mesh) using ethyl acetate-hexane (30:70) as eluent. The solvents were removed under reduced pressure to give the product. White powder; m.p. 67-69 ºC, yield 85 %; IR (KBr, νmax, cm-1): 1745, 1735 and 1710 (C=O), 1627 (C=C); 1H-NMR (CDCl3): δH 1.94 (2H, t, 3JHH = 7.4 Hz, CH2), 1.98 (3H, s, CH3), 2.50 (2H, t, 3JHH = 7.3 Hz, CH2), 2.70 (2H, m, CH2), 3.66 (3H, s, OCH3), 3.73 (3H, s, OCH3), 6.71 (1H, s, CH); 13C-NMR (CDCl3): δC 19.58 and 22.61 (2CH2), 28.42 (CH3), 39.06 (CH2), 51.78 and 52.67 (2OCH3), 124.89, 134.74, 139.82 and 149.97 (olefinic carbons), 165.06 and 165.22 (2C=O, esters), 206.29 (C=O, ketone); MS m/z (%): 252 (M+, 21), 237 (M+- Me, 25), 221 (M+-OMe, 34), 193 (M+-CO2Me, 42), 162 [M+-(CO2Me+OMe), 48], 110 (M+-MeO2CCCCO2Me, 100); Anal. calcd. for C13H16O5 (252.27): C 61.90, H 6.39 %; Found C 61.77, H 6.30 %. The following compounds were prepared similarly:
Diethyl (E)-2-[1-(2-oxocyclopentylidene)ethyl]-2-butenedioate (5b). White powder; m.p. 66-68 ºC; yield 80 %; IR (KBr, νmax, cm-1): 1740, 1730 and 1710 (C=O), 1620 (C=C); 1H-NMR (CDCl3): δH 1.25-1.44 (6H, m, 2CH3), 2 (2H, t, 3JHH = 7.8 Hz, CH2), 2.04 (3H, s, CH3), 2.30-2.40 (2H, t, 3JHH = 7.7 Hz, CH2), 2.75-2.77 (2H, m, CH2), 4.17 (2H, q, 3JHH = 7.1 Hz, OCH2), 4.25 (2H, q, 3JHH = 7.1 Hz, OCH2), 6.77 (1H, s, CH); 13C-NMR (CDCl3): δC 14.12 and 14.14 (2CH3), 19.62 and 22.16 (2CH2), 28.45 (CH3CO), 39.08 (CH2), 60.07 and 61.60 (2OCH2), 125.18, 134.55, 140.1 and 149.92 (olefinic carbons), 164.57 and 164.9 (2C=O, esters), 203.5 (C=O, ketone); ); MS m/z (%): 280 (M+, 16), 265 (M+-Me, 22), 207 (M+-CO2Et, 28), 162 [M+-(CO2Et+OEt), 48], 110 (M+-EtO2CCCCO2Et, 100); Anal. calcd. for C15H20O5 (280.32): C 64.27, H 7.19 %; Found C 64.15, H 7.08 %.
Di-tert-butyl (E)-2-[1-(2-oxocyclopentylidene)ethyl]-2-butenedioate (5c). White powder; m.p. 64-67 ºC; yield 87 %; IR (KBr, νmax, cm-1): 1738, 1730 and 1715 (C=O), 1618 (C=C); 1H-NMR (CDCl3): δH 1.40 and 1.47 (2CMe3), 1.92 (2H, t, 3JHH = 7.4 Hz, CH2), 1.95 (3H, s, CH3CO), 2.22-2.27 (2H, t, 3JHH = 7.3 Hz, CH2), 2.65-2.70 (2H, m, CH2), 6.54 (1H, s, CH); 13C-NMR (CDCl3): δC 19.62 and 22.44 (2CH2), 27.92 and 27.98 (2CMe3), 28.48 (CH3), 39.10 (CH2), 81.02 and 81.82 (2CMe3), 126.60, 133.94, 140.63 and 149.52 (olefinic carbons), 163.86 and 164.43 (2C=O, esters), 205.71 (C=O, ketone); MS m/z (%): 336 (M+, 8), 280 (M+- C4H8, 22), 263 (M+-OtBu, 28), 235 (M+-CO2tBu, 20), 179 [M+-(CO2tBu+C4H8), 35], 162 [M+-(CO2tBu+OtBu), 42], 110 (M+-tBuO2CCCCO2tBu, 54); Anal. calcd. for C19H28O5 (336.43): C 67.83, H 8.38 %; Found C 67.70, H 8.26 %.

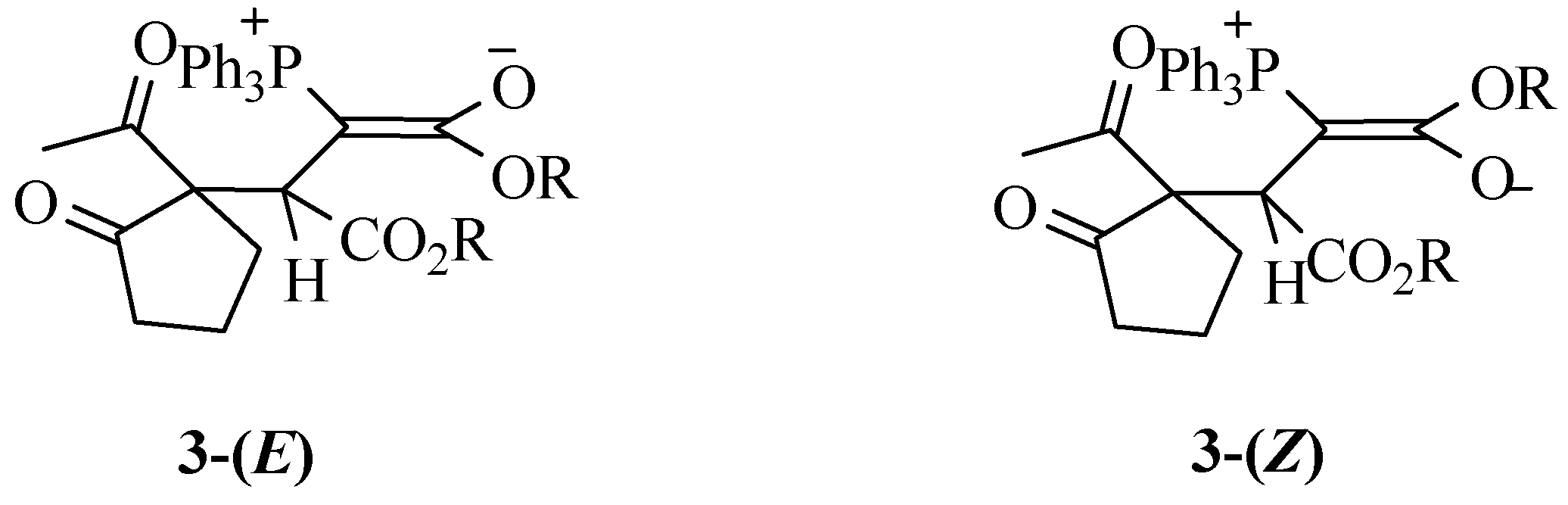{kind=link}
{kind=link}
{kind=link}
{kind=link}



