Results and Discussion
The starting 6-bromo-1,4-dimethyl-9
H-carbazole (
4) was prepared by a known procedure [
13,
14]. Classic cross-coupling conditions (Na
2CO
3/Pd(PPh
3)
4/1,4-dioxane/water/80°C) [
6] applied to the reaction of this compound with arylboronic acids
I and
II proved to be ineffective for the production of the desired 6-aryl-1,4-dimethyl-9
H-carbazoles
5 (
Scheme 1).
Scheme 1.
Cross-coupling reaction with 6-bromo-1,4-dimethyl-9H-carbazole 4.
Scheme 1.
Cross-coupling reaction with 6-bromo-1,4-dimethyl-9H-carbazole 4.
Reagents: (i) ArB(OH)2 (I and II), 1,4-dioxane/H2O, Na2CO3, Pd(PPh3)4, 80°C, 48h.
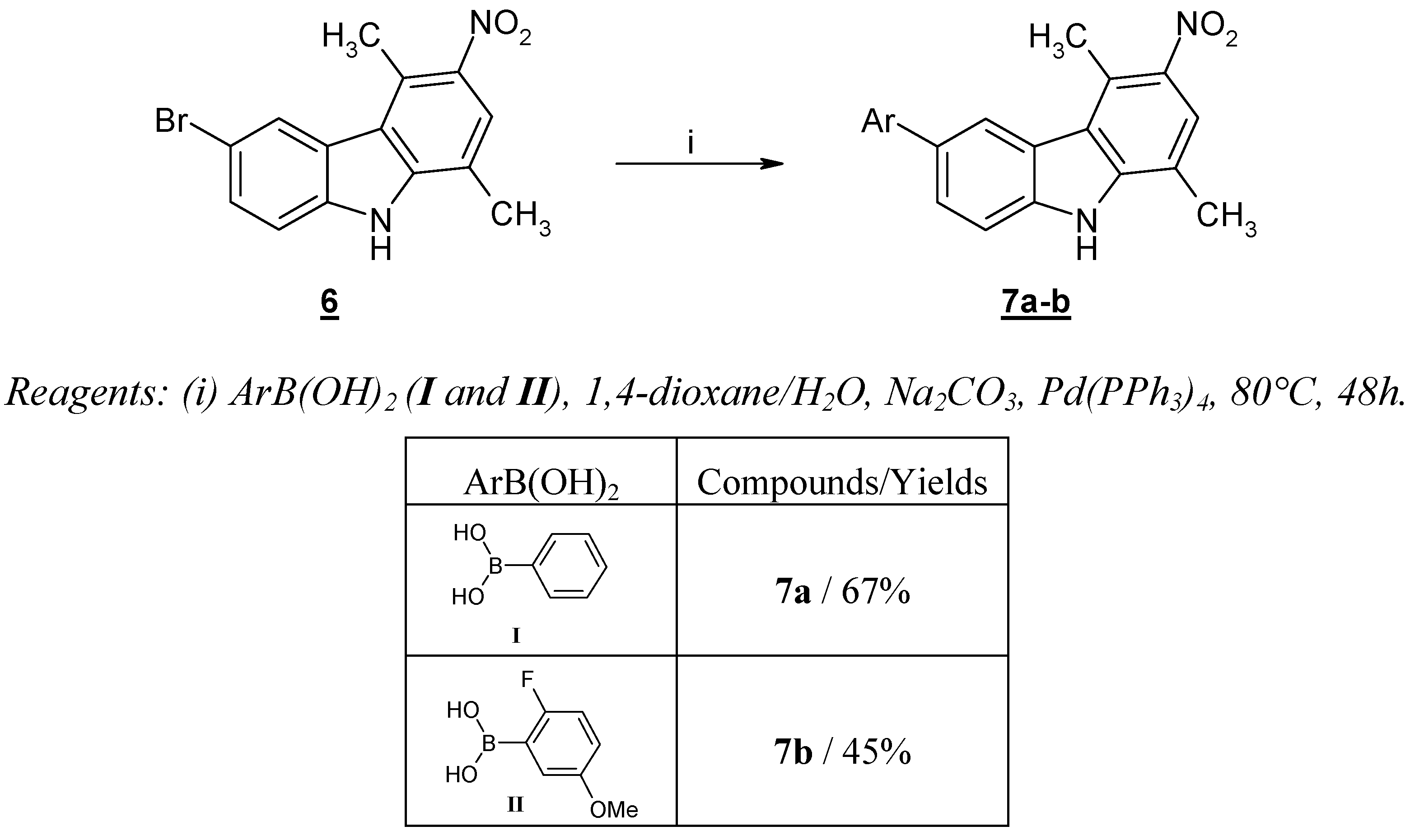
However a similar experimental procedure was effective when applied to the reaction of arylboronic acids
I and
II and 6-bromo-1,4-dimethyl-3-nitro-9
H-carbazole (
6), giving aryl-1,4-dimethyl-3-nitro-9
H-carbazoles
7a and
7b, in reasonable yields (
Scheme 2). As already described in numerous cases, the presence of a strong electron-withdrawing group favored the success of the reaction [
15].
Scheme 2.
Cross-coupling reaction with 6-bromo-1,4-dimethyl-3-nitro-9H-carbazole (6).
Scheme 2.
Cross-coupling reaction with 6-bromo-1,4-dimethyl-3-nitro-9H-carbazole (6).
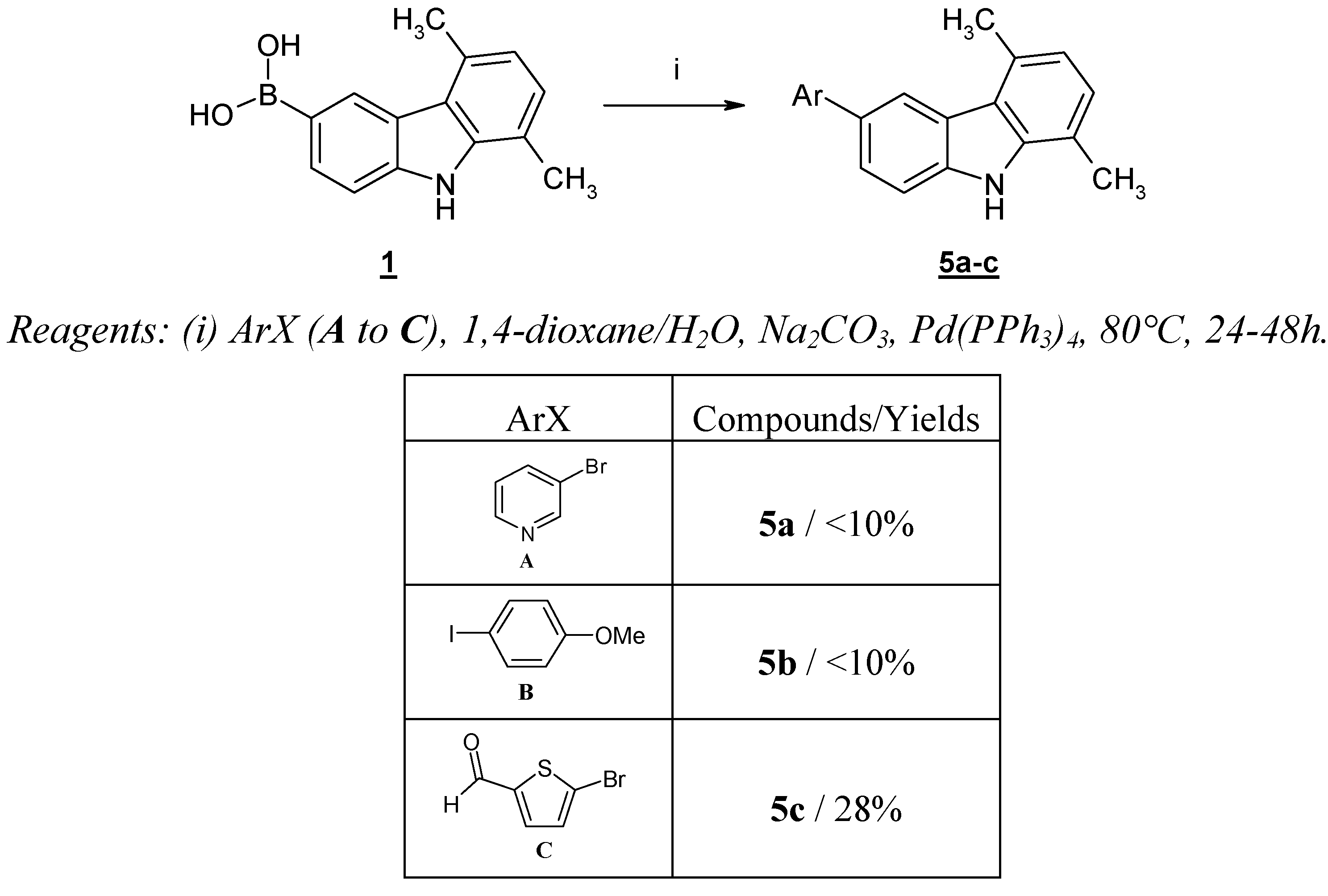
Faced with these difficulties and also because the nitro group was considered undesirable, we have studied the reverse strategy, consisting in the preparation of the boronic acid
1. We have already described the synthesis of this
N-unprotected boronic acid
1 [
9] starting from 6-bromo-1,4-dimethyl-9
H-carbazole (
4).
Scheme 3.
Cross-coupling reaction with 5,8-dimethyl-9H-carbazole-3-boronic acid (1).
Scheme 3.
Cross-coupling reaction with 5,8-dimethyl-9H-carbazole-3-boronic acid (1).
Unfortunately, this boronic acid is a very poor partner in cross-coupling reactions and yields obtained with (hetero)aryl halides such as 3-bromopyridine (
A) and
p-iodoanisole (
B) remained very modest (<10%), the exception being a heteroaryl halide bearing an electron withdrawing group such as 5-bromo-2-thiophenecarboxaldehyde (
C), which gave the product
5c in 28% yield (
Scheme 3).
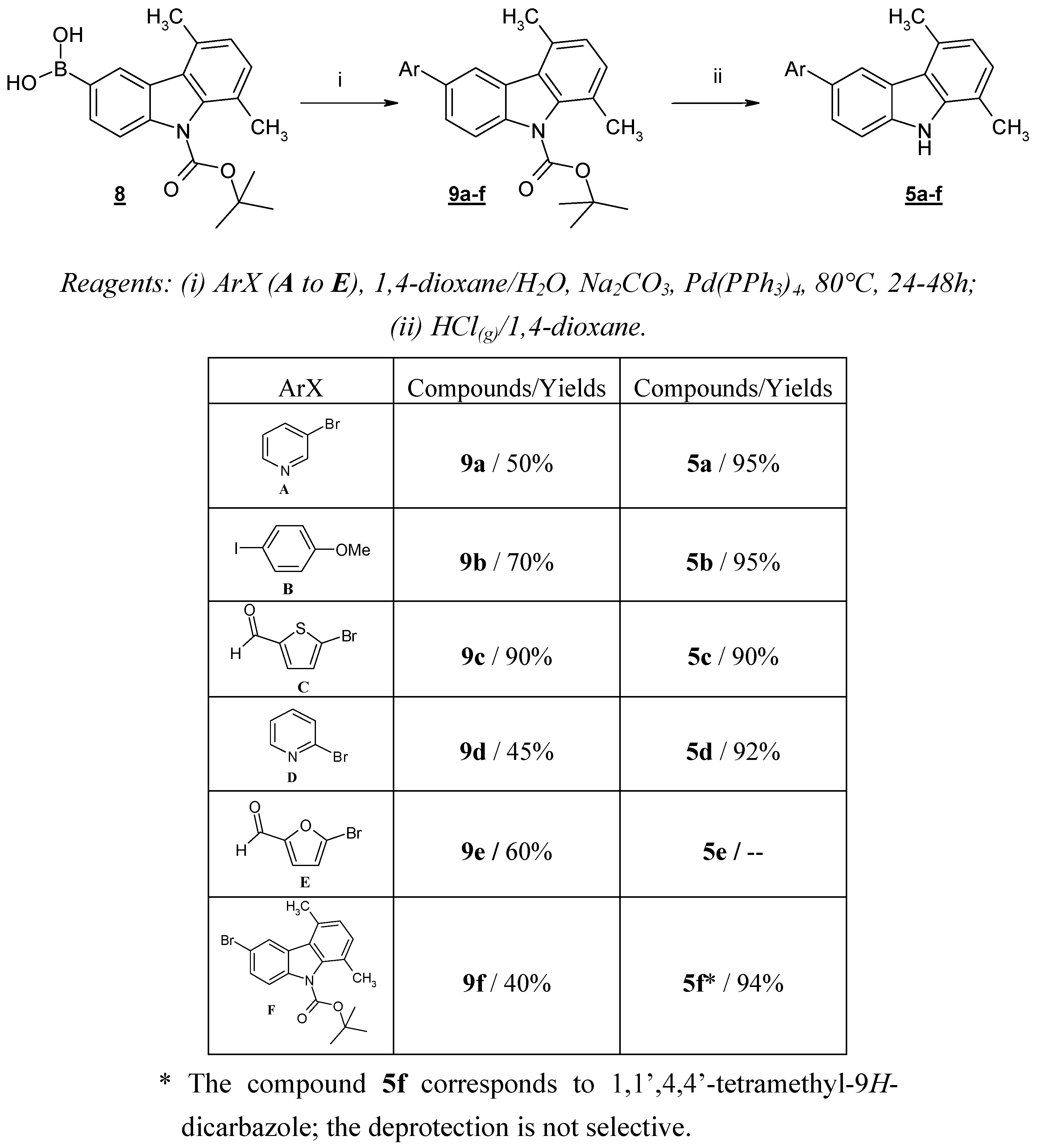
These failures prompted us to protect the nitrogen atom of 5,8-dimethyl-9
H-carbazole-3-boronic acid (
1). Thus, we prepared
N-Boc-5,8-dimethyl-9
H-carbazol-3-boronic acid (
8), which was obtained after
N-Boc-protection and successive lithiation-boronation of compound
4. We previously described the use of this reaction sequence for the preparation of 5,8-dimethyl-9
H-carbazole-3-ol [
9].
Scheme 4.
Cross-coupling reaction with N-Boc-5,8-dimethyl-9H-carbazol-3-boronic acid (8).
Scheme 4.
Cross-coupling reaction with N-Boc-5,8-dimethyl-9H-carbazol-3-boronic acid (8).
Compound
8 was then subjected to the Suzuki cross-coupling reaction with a variety of (hetero)aryl halides under standard conditions [
6]. An aqueous solution of Na
2CO
3 and Pd(PPh
3)
4 was slowly added to a mixture of compound
8 and the appropriate (hetero)aryl halide in 1,4-dioxane at room temperature. The resulting mixture was then heated to reflux for the given time. After the usual work-up, compounds
9a-f were obtained in good yields. The
N-deprotection of
9a-
f was carried out with gaseous HCl in 1,4-dioxane, giving compounds
5a,
5b,
5c,
5d and
5f in quantitative yields (
Scheme 4). We were unable to isolate compound
5e, probably due to the instability of the furfural moiety in an acidic medium.
Experimental
General
Commercial reagents were purchased from Aldrich, Acros Organics or Alfa Aesar and used without additional purification. Melting points were determined on a Kofler melting point apparatus. IR spectra were taken with a Perkin Elmer BX FT-IR. Mass spectra were taken on a JEOL JMS GCMate spectrometer at ionizing potential of 70 eV (EI) or were performed using a LC-MS Waters Alliance 2695 spectrometer (ESI+). 1H-NMR (400 MHz) was recorded on a JEOL Lambda 400 spectrometer; 13C-NMR were not recorded due to the low solubility of the compounds. Chemical shifts are expressed in parts per million downfield from tetramethylsilane as an internal standard. Thin layer chromatography (TLC) was performed on silica gel 60F-264 (Merck).
General procedure for the preparation of compounds 7a-b
The preparation of 1,4-dimethyl-3-nitro-6-phenyl-9H-carbazole (7a) is reported as a representative example: to a solution of 6-bromo-1,4-dimethyl-3-nitro-9H-carbazole (6, 1.50 g, 4.70 mmol) in 1,4-dioxane (70 mL), kept under an argon atmosphere, phenylboronic acid (I, 0.63 g, 5.17 mmol), Na2CO3 (1.25 g, 11.79 mmol) in H2O (3.50 mL) and Pd(PPh3)4 (0.30 g, 0.26 mmol) were added sequentially. The reaction mixture was heated to reflux for 48h, and then volatiles were evaporated under reduced pressure. The solid formed was recrystallized from acetonitrile to give 1,4-dimethyl-3-nitro-6-phenyl-9H-carbazole (7a). Yellow solid (67 % yield), mp = 226 °C; IR (KBr): 3451, 1584, 1521, 1302, 1301, 817, 758 cm-1; 1H-NMR (DMSO-d6) δ 11.94 (s, 1H, NH); 8.35 (s, 1H, H2); 7.86 (s, 1H, H5); 7.83-7.83 (m, 7H, H7, H8, C6H5); 2.96 (s, 3H, CH3); 2.55 (s, 3H, CH3); MS (EI) m/z (%): 316 (M+ •, 98), 271 (48) (M+ •-NO2).
1,4-Dimethyl-3-nitro-6-(2’-fluoro-5'-methoxyphenyl)-9H-carbazole (7b)
Prepared following the general procedure but employing 2-fluoro-5-methoxyphenylboronic acid (II) in place of I. Yellow solid (45 % yield), mp = 240 °C; IR (KBr): 3309, 1578, 1299, 1205, 1034, 810, 756 cm-1; 1H-NMR (DMSO-d6) δ 12.06 (s, 1H, NH); 8.36 (s, 1H, H2); 7.87 (s, 1H, H5); 7.71-7.65 (m, 2H, H8, H6’); 7.28-7.23 (m, 1H, H7); 7.17-7.14 (m, 1H, H4’); 6.95-6.93 (m, 1H, H3’); 3.80 (s, 3H, OCH3); 2.99 (s, 3H, CH3); 2.58 (s, 3H, CH3); MS (ESI+): 364 (M++1).
General procedure for the preparation of compounds 9a-f
The preparation of 1,4-dimethyl-6-pyridin-3-yl-9-tert-butoxycarbonyl-9H-carbazole (9a) is reported as a representative example: to a solution of 3-bromopyridine (A, 0.08 mL, 0.80 mmol) in 1,4-dioxane (50 mL), kept under an argon atmosphere, 9-tert-butoxycarbonyl-5,8-dimethyl-9H-carbazole-3-boronic acid (8, 0.30 g, 0.88 mmol), Na2CO3 (0.21 g, 2 mmol) in H2O (2 mL) and Pd(PPh3)4 (0.05 g, 0.04 mmol) were added sequentially. The reaction mixture was heated to reflux for 48 h, and then concentrated under reduced pressure. The solid residue was recrystallized from acetonitrile. Compound 9a was obtained as a light brown solid (50 % yield), mp = 167 °C; IR (KBr): 3435, 2972, 1731, 1458, 1288, 1246, 1145, 1084, 809, 721, 619 cm-1; 1H-NMR (DMSO-d6) δ 9.01 (s, 1H, H2’); 8.60-8.56 (m, 1H, H6’); 8.35 (s, 1H, H5); 8.20 (d, JH4’-5’ = 7.8 Hz, 1H, H4’); 8.11 (d, JH7-8 = 8.8 Hz, 1H, H7); 7.87 (d, JH8-7 = 8.8 Hz, 1H, H8); 7.53-7.50 (m, 1H, H5’); 7.24 (d, JH2-3 = 7.8 Hz, 1H, H2); 7.15 (d, JH3-2 = 7.8 Hz, 1H, H3); 2.84 (s, 3H, CH3); 2.40 (s, 3H, CH3); 1.67 (s, 9H, 3CH3); MS (EI) m/z (%): 372 (M+ •, 1), 316 (100) (M+ •- tBu), 272 (100) (M+ •- CO2tBu). MS (ESI+): 373 (M++1).
6-(4’-Methoxy-phenyl)-1,4-dimethyl-9-tert-butoxycarbonyl-9H-carbazole (9b)
Following a procedure identical to that described for 9a, but using 4-iodoanisole (B, 0.30 g, 1.34 mmol), 9b was obtained as a white solid (70 % yield), mp = 122 °C; IR (KBr): 2977, 1728, 1451, 1285, 1246, 1150, 1081, 804, 757, 697 cm-1; 1H-NMR (DMSO-d6) δ 8.36 (s, 1H, H5); 8.15 (d, JH7-8 = 8.8 Hz, 1H, H7); 7.89-7.83 (m, 2H, H8, H3’); 7.58-7.55 (m, 2H, H2’, H6’); 7.46-7.44 (m, 1H, H5’); 7.29 (d, JH2-3 = 7.8 Hz, 1H, H2); 7.20 (d, JH3-2 = 7.8 Hz, 1H, H3); 3.47 (s, 3H, OCH3); 2.89 (s, 3H, CH3); 2.45 (s, 3H, CH3); 1.71 (s, 9H, 3CH3); MS (EI) m/z (%): 401 (M+ •, 1), 271 (100) (M+ •- OCH3 - CO2tBu).
5-(5’,8’-Dimethyl-9’-tert-butoxycarbonyl-9’H-carbazol-3’-yl)-thiophene-2-carbaldehyde (9c)
Following a procedure identical to that described for 9a, but using 5-bromo-2-thiophene-carboxaldehyde (C, 0.27 g, 1.54 mmol) and a reflux time of 24 h, compound 9c was obtained as a yellow solid (90 % yield), mp = 168 °C; IR (KBr): 3428, 2971, 1739, 1657, 1428, 1225, 1149, 1061, 797, 665 cm-1; 1H-NMR (DMSO-d6) δ 9.90 (s, 1H, CHO); 8.38 (s, 1H, H4’) 8.07-8.05 (m, 2H, H1’, H3); 7.94 (d, J2-1 = 8.8 Hz, 1H, H2); 7.82 (d, JH4-3 = 3.9 Hz, 1H, H4); 7.24 (d, JH7-6 = 7.8 Hz, 1H, H7); 7.15 (d, JH6-7 = 7.8 Hz, 1H, H6); 2.81 (s, 3H, CH3); 2.40 (s, 3H, CH3); 1.66 (s, 9H, 3CH3); MS (ESI+): 405 (M++1).
1,4-Dimethyl-6-pyridin-2-yl-9-tert-butoxycarbonyl-9H-carbazole (9d)
Following a procedure identical to that described for 9a, but using 2-bromopyridine (D, 0.09 mL, 0.98 mmol), 9d was obtained as a light brown solid (45 % yield), mp = 212 °C; IR (KBr): 3370, 2977, 1701, 1457, 1290, 1252, 1149, 1088, 803, 780, 670 cm-1; 1H-NMR (DMSO-d6) δ 8.66 (s, 1H, H5); 8.08 (d, JH7-8 = 8.8 Hz, 1H, H7); 8.07-7.95 (m, 1H, H6’); 7.75 (d, JH8-7 = 8.8 Hz, 1H, H8); 7.71-7.68 (m, 2H, H4’, H3’); 7.20-715 (m, 1H, H5’); 7.10 (d, JH2-3 = 7.8 Hz, 1H, H2); 7.02 (d, JH3-2 = 7.8 Hz, 1H, H3); 2.72 (s, 3H, CH3); 2.41 (s, 3H, CH3); 1.62 (s, 9H, 3CH3); MS (ESI+): 373 (M++1).
5-(5’,8’-Dimethyl-9’-tert-butoxycarbonyl-9’H-carbazol-3’-yl)-furan-2-carbaldehyde (9e)
Following a procedure identical to that described for 9a, but using 5-bromo-2-furaldehyde (E, 0.27 g, 1.54 mmol) and a reflux time of 24 h, compound 9e was obtained as a yellow solid (60 % yield), mp = 110 °C; IR (KBr): 3430, 2975, 1744, 1672, 1450, 1285, 1149, 1084, 795 cm-1; 1H-NMR (DMSO-d6) δ 9.62 (s, 1H, CHO); 8.48 (s, 1H, H4’) 8.10 (d, JH1’-2’ = 8.8 Hz, 1H, H1’); 8.03 (d, JH2’-1’ = 8.79 Hz, 1H, H2’); 7.67 (d, JH3-4 = 3.9 Hz, 1H, H3); 7.34 (d, JH4-3 = 3.9 Hz, 1H, H4); 7.25 (d, JH7-6 = 7.8 Hz, 1H, H7); 7.16 (d, JH6-7 = 7.8 Hz, 1H, H6); 2.82 (s, 3H, CH3); 2.42 (s, 3H, CH3); 1.67 (s, 9H, t-Bu); MS (ESI+): 390 (M++1).
1,1’,4,4’-Tetramethyl-9-tert-butoxycarbonyl-9H-dicarbazole (9f)
Following the identical procedure to that described for 9f, but using 6-bromo-1,4-dimethyl-9-tert-butoxycarbonyl-9H-carbazole (F, 0.30 g, 0.80 mmol), 9f was obtained as a white solid (40 % yield), mp = 214 °C; IR (KBr): 3439, 2979, 1726, 1453, 1295, 1248, 1154, 1083, 804, 541 cm-1; 1H-NMR (DMSO-d6) δ 8.40 (s, 2H, H5, H5’); 8.20-8.10 (m, 2H, H7, H7’); 8.00-7.90 (m, 2H, H8, H8’); 7.10-7.30 (m, 4H, H2, H3, H2’, H3’); 2.85 (s, 6H, 2CH3); 2.42 (s, 6H, 2CH3); 1.68 (s, 18H, 6CH3); MS (EI) m/z (%): 588 (M+ •, 1), 388 (100) (M+ • - 2 CO2tBu).
General procedure for the deprotection of compounds 9a-f
The preparation of 1,4-dimethyl-6-pyridin-3-yl-9H-carbazole (5a) is reported as a representative example: to a mixture of 1,4-dioxane (70 mL) and gaseous HCl (saturated solution) cooled to 0 °C compound 9a (2.36 mmol) was added. The reaction mixture was heated to reflux for 48 h, then concentrated under reduced pressure. The oil formed residue was crystallized from acetonitrile. Compound 5a was obtained as a yellow solid (95 % yield), mp > 260 °C; IR (KBr): 3426, 2518, 1470, 1258, 790, 671 cm-1; 1H-NMR (DMSO-d6) δ 11.51 (s, 1H, NH); 9.30 (d, JH2’4’ = 2.4 Hz, 1H, H2’); 8.84 (dd, JH6’-5’ = 4.4 Hz, JH6’-4’ = 1.2 Hz, 1H, H6’); 8.77 (part A of AB system, JAB = 8.3 Hz, JHA-2’ = 2.4 Hz, JHA-6’ = 1.2 Hz, 1H, H4’); 8.49 (s, 1H, H5); 8.03 (part B of AB system, JAB = 8.3 Hz, JHB-6’ = 4.4 Hz, 1H, H5’); 7.85 (d, JH7-8 = 8.8 Hz, 1H, H7); 7.68 (d, JH8-7 = 8.8 Hz, 1H, H8); 7.13 (d, JH2-3 = 6.8 Hz, 1H, H2); 6.91 (d, JH3-2 = 6.8 Hz, 1H, H3); 3.42 (1H, NH+Cl-); 2.85 (s, 3H, CH3); 2.52 (s, 3H, CH3); MS (ESI+): 273 (M++1).
The following compounds were similarly obtained:
6-(4’-Methoxyphenyl)-1,4-dimethyl-9H-carbazole (5b). White solid (95 % yield), mp = 200 °C; IR (KBr): 3020, 1454, 1320, 1250, 1170, 1076, 799, 770, 676 cm-1; 1H-NMR (DMSO-d6) δ 10.00 (s, 1H, NH); 7.85 (s, 1H, H5); 7.58-7.56 (m, 2H, H2’, H6’); 7.40 (d, JH8-7 = 8.8 Hz, 1H, H8); 7.30 (d, JH7-8 = 8.8 Hz, 1H, H7); 7.05-7.00 (m, 2H, H3’, H5’); 6.98 (d, JH2-3 = 7.8 Hz, 1H, H2); 6.89 (d, JH3-2 = 7.8 Hz, 1H, H3); 3.85 (s, 3H, OCH3); 2.84 (s, 3H, CH3); 2.54 (s, 3H, CH3); MS (EI) m/z (%): 301 (M+ •, 100).
5-(5’,8’-Dimethyl-9’H-carbazol-3’-yl)-thiophene-2-carbaldehyde (5c). Yellow solid (90 % yield), mp = 262 °C; IR (KBr): 3435, 3267, 1634, 1430, 1241, 1058, 800, 669 cm-1; 1H-NMR (DMSO-d6) δ 11.58 (s, 1H, NH); 9.94 (s, 1H, CHO); 8,50 (s, 1H, H4); 8.11-8.10 (d, J3’‑4’= 3.9 Hz 1H, H3’); 7.89 (d, J2-1= 8.8 Hz 1H, H2); 7.83 (d, JH4’-3’ = 3.9 Hz 1H, H4’); 7.67 (d, J1-2 = 8.8 Hz 1H, H1); 7.19 (d, JH7-6 = 7.8 Hz, 1H, H7); 6.97 (d, JH6-7= 7.8 Hz, 1H, H6); 2.89 (s, 3H, CH3); 2.58 (s, 3H, CH3); MS (ESI+): 306 (M++1).
1,4-Dimethyl-6-pyridin-2yl-9H-carbazole (5d). White solid (92 % yield), mp > 260 °C; IR (KBr): 3424, 2516, 1468, 1270, 800, 673 cm-1; 1H-NMR (DMSO-d6) δ 11.48 (s, 1H, NH); 8.64 (s, 1H, H5); 8.10 (d, JH7-8 = 8.8 Hz, 1H, H7); 8.09-7.92 (m, 1H, H6’); 7.79 (d, JH8-7 = 8.8 Hz, 1H, H8); 7.70-7.69 (m, 2H, H4’, H3’); 7.22-718 (m, 1H, H5’); 7.13 (d, JH2-3 = 7.8 Hz, 1H, H2); 7.07 (d, JH3-2 = 7.8 Hz, 1H, H3); 3.43 (1H, NH+Cl-); 2.73 (s, 3H, CH3); 2.45 (s, 3H, CH3); MS (ESI+): 273 (M++1).
1,1’,4,4’-Tetramethyl-9H-dicarbazole (5f). White solid (94 % yield), mp = 254 °C; IR (KBr): 3454, 2987, 1457, 1300, 1254, 1187, 1067, 812, 570 cm-1; 1H-NMR (DMSO-d6) δ 10.20 (s, 2H, NH); 7.99-7.94 (m, 2H, H5, H5’); 7.59-7.38 (m, 4H, H7, H8, H7’, H8’); 7.15-6.98 (m, 4H, H2, H3, H2’, H3’); 2.85 (s, 6H, 2CH3); 2.55 (s, 6H, 2CH3); MS (EI) m/z: 388 (M+ •, 100).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





