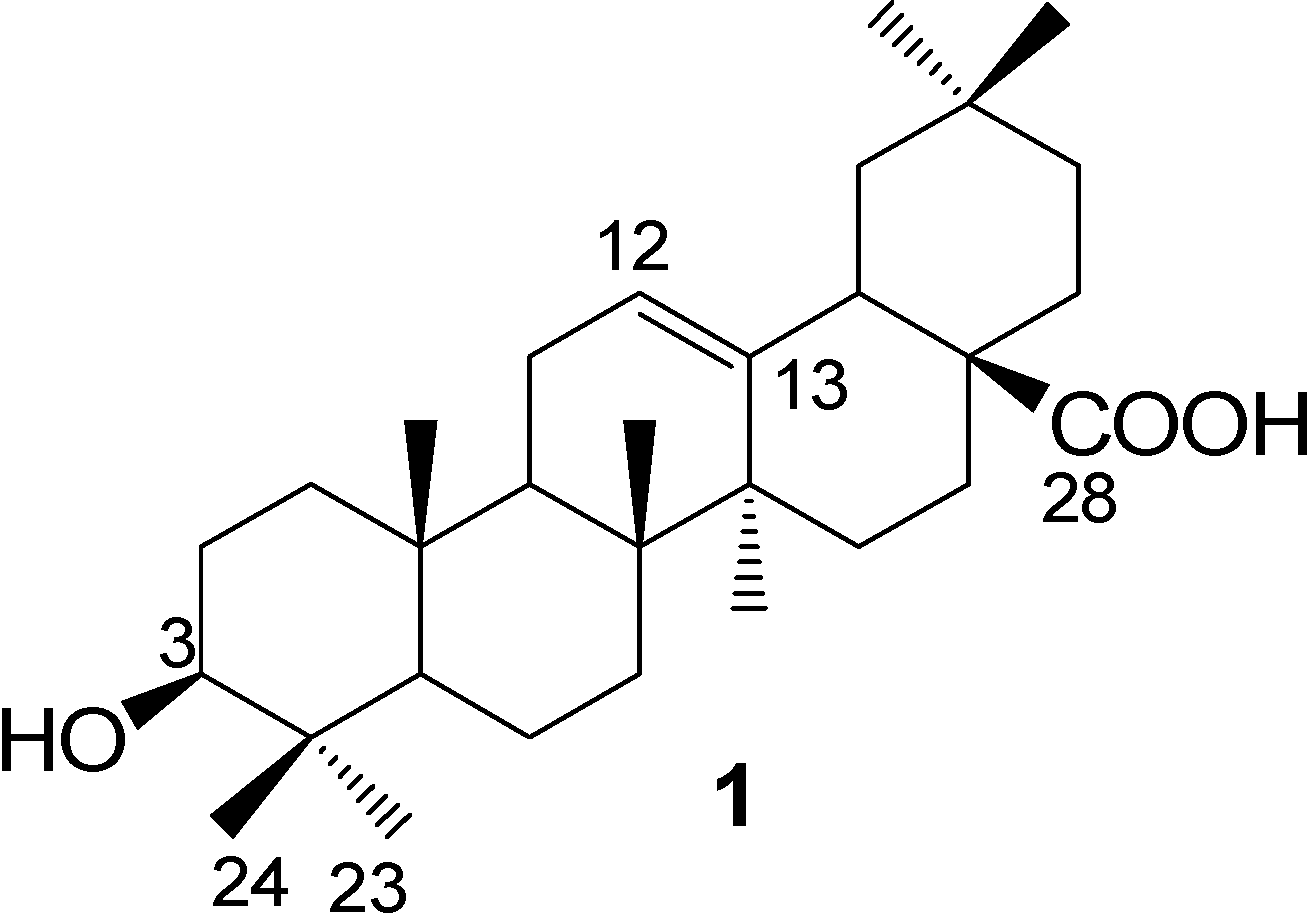
Experimental
General
Commercial reagents were used without further treatment unless specialized. Solvents were dried and distilled prior to use in the usual way. Boiling range of petroleum ether was 60~90°C. Analytical TLC was performed with silica gel GF
254. Preparation column chromatography was performed with silica gel H. Saccharide donors (trichloroacetimidates and bromoglycosides) were prepared according to the reported methods [
16,
25,
26].
1H-NMR and
13C-NMR spectra were recorded on a Bruker ARX 300 MHz instrument.
J values were given in Hz. ESI-MS were obtained on an Agilent 1100 mass spectrometer. HRMS was detected on High resolution ESI-FTICR mass spectrometry (Ion spec 7.0T).
Synthesis of OA 3-glycosides 6a~6g
Benzyl oleanolate (2).
A suspension of OA (5.00 g, 10.9 mmol), BnBr (2.10 mL, 17.5 mmol) and K
2CO
3 (3.00 g, 21.8 mmol) in THF-H
2O (40:1, 82 mL) was stirred overnight at rt. The mixture was then filtered, and the filtrate was concentrated under vacuum and purified through a silica gel column chromatography (8:1, petroleum ether-EtOAc) to give
2 (5.84 g, 98%) as a white amorphous solid. The physical data agreed with that previously reported [
19].
Oleanolic acid 3-O-α-L-arabinopyranoside (6d)
Benzyl ester 2 (200 mg, 0.366 mmol), trichloroacetimidate 3d (244 mg, 0.402 mmol) and powdered 4 Å molecular sieves (500 mg) were stirred for 30 min at rt in dry CH2Cl2 (6 mL). A solution of TMSOTf in dry CH2Cl2 (1%, 0.35 mL) was added dropwise. The mixture was stirred for 20 min followed by addition of Et3N (0.10 mL) and filtration. The filtrate was concentrated and purified by a silica gel column chromatography (8:1, petroleum ether-EtOAc) to afford benzyl oleanolate 3-O-(2,3,4-tri-O-benzoyl-α-L-arabinopyranoside) (4d) (330 mg, 91%) as a white amorphous solid. Rf = 0.65 (4:1, petroleum ether-EtOAc); 1H-NMR (CDCl3) δ 8.09-7.26 (m, 20H, H-C(Ar)), 5.75 (dd, J=8.8, 6.5, 1H, H-C(2')), 5.67 (m, 1H, H-C(4')), 5.59 (dd, J=8.9, 3.5, 1H, H-C(3')), 5.30 (t, J=3.0, 1H, H-C(12)), 5.06 (dd, J=19.9, 12.6, 2H, PhCH2), 4.79 (d, J=6.6, 1H, H-C(1')), 4.33 (dd, J=12.9, 3.8, 1H, H-C(5')-1), 3.87 (m, 1H, H-C(5')-2), 3.16 (dd, J=11.0, 4.9, 1H, H-C(3)), 2.90 (dd, J=10.1, 3.2, 1H, H-C(18)), 1.11, 0.92, 0.90, 0.86, 0.79, 0.66, 0.58 (s, 7×3H, CH3); ESI-MS: 1013.5 [(M+Na)+].
A suspension of 4d (200 mg, 0.202 mmol) and 10% Pd-C (32 mg) in EtOAc (8 mL) was refluxed and bubbled up with H2 (20 mL/min) for 3 h. The mixture was then filtered and the filtrate was concentrated to dryness to afford oleanolic acid 3-O-(2,3,4-tri-O-benzoyl-α-L-arabinopyranoside) (5d) (180 mg, 99%) as a white amorphous solid. Rf = 0.36 (3:1, petroleum ether-EtOAc); 1H-NMR (CDCl3) δ 8.07-7.18 (m, 15H, H-C(Ar)), 5.77 (dd, 1H, J=8.8, 6.4, H-C(2')), 5.67 (m, 1H, H-C(4')), 5.59 (dd, 1H, J=8.8, 3.5, H-C(3')), 5.26 (br s, 1H, H-C(12)), 4.78 (d, 1H, J=6.3, H-C(1')), 4.33 (dd, 1H, J=13.0, 3.9, H-C(5')-1), 3.87 (dd, 1H, J=13.0, 1.9, H-C(5')-2), 3.16 (dd, 1H, J=11.1, 4.8, H-C(3)), 2.80 (dd, 1H, J=9.9, 2.6, H-C(18)), 1.10, 0.92, 0.90, 0.87, 0.77, 0.70, 0.62 (s, 7×3H, CH3); ESI-MS: 918.8 [(M+NH4)+].
Compound 5d (84 mg, 0.093 mmol) was suspended in dry MeOH (5 mL), to which a freshly prepared solution of NaOMe in MeOH (1.0 M, 0.40 mL) was added. The mixture was stirred at rt overnight and then neutralized with Dowex H+ resin to pH 7 and filtered. The filtrate was concentrated and purified with a silica gel column chromatography (9:1, CHCl3-MeOH) to give oleanolic acid 3-O-α-L-arabinopyranoside (6d) (47 mg, 85%) as a white powder. Rf = 0.56 (5:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 5.47 (br s, 1H, H-C(12)), 4.77 (d, 1H, J=7.0, H-C(1')), 4.43 (t, 1H, J=8.7), 4.31 (m, 2H), 4.16 (dd, 1H, J=8.7, 3.1), 3.83 (m, 1H), 3.37-3.27 (m, 2H, H-C(3), H-C(18)), 1.29, 1.27, 1.00, 0.99, 0.94, 0.93, 0.83 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 180.2, 144.8, 122.6, 107.6, 88.7, 74.7, 73.0, 69.6, 66.9, 55.9, 48.1, 46.7, 46.5, 42.2, 42.0, 39.8, 39.6, 38.8, 37.0, 34.2, 33.3, 33.2, 33.2, 31.0, 28.2, 28.2, 26.7, 26.2, 23.8, 23.8, 23.7, 18.5, 17.4, 16.9, 15.5; ESI-MS: 611.5 [(M+Na)+], 627.3 [(M+K)+]. HRMS: m/z 587.3962 [M-H]- ([C35H55O7] = 587.3953).
Compounds 6a~6c and 6e~6g were prepared according to the same procedure described for 6d.
Oleanolic acid 3-O-β-D-glucopyranoside (6a). Rf = 0.39 (5:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 5.44 (br s, 1H, H-C(12)), 4.93 (d, 1H, J=7.7, H-C(1')), 4.57 (br d, 1H, J=11.3), 4.43 (m, 1H), 4.25 (m, 2H), 4.01 (m, 2H), 3.46 (dd, 1H, J=12.5, 2.2, H-C(18)), 3.37 (dd, 1H, J=8.0, 2.4, H-C(3)), 1.31, 1.30, 1.01, 0.98, 0.93, 0.91, 0.80 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 180.2, 145.4, 122.4, 107.0, 88.8, 78.8, 78.5, 78.4, 71.8, 63.0, 55.8, 48.0, 46.8, 46.7, 42.2, 42.1, 39.7, 39.5, 38.7, 37.0, 34.4, 33.4, 33.3, 33.0, 31.1, 30.0, 29.9, 28.3, 26.2, 23.9, 23.9, 23.8, 18.5, 17.5, 17.1, 15.5; ESI-MS: 641.4 [(M+Na)+], 653.4 [(M+Cl)-]. HRMS: m/z 617.4071 [M-H]- ([C36H57O8] = 617.4059).
Oleanolic acid 3-O-β-D-galactopyranoside (6b). Rf = 0.40 (5:1, CHCl3-MeOH); 1H-NMR (DMSO-d6) δ12.07 (s, 1H, COOH), 5.16 (br s, 1H, H-C(12)), 4.75, 4.64, 4.52, 4.33 (br s, 4×1H, OH), 4.11 (d, 1H, J=6.4, H-C(1')), 3.62 (br s, 1H), 3.52 (m, 1H), 3.43 (m, 1H), 3.29-3.25 (m, 3H), 3.03 (br d, J=7.8, H-C(3)), 2.74 (br d, 1H, J=10.6, H-C(18)), 1.10 (s, 3H, CH3), 0.98 (s, 3H, CH3), 0.88 (s, 9H, 3×CH3), 0.76 (s, 3H, CH3), 0.72 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 178.7, 143.9, 121.6, 106.2, 88.0, 75.0, 73.7, 71.2, 68.2, 60.4, 55.1, 47.2, 45.8, 45.6, 41.4, 40.9, 38.7, 38.3, 38.2, 36.4, 33.4, 32.9, 32.5, 32.2, 30.5, 27.8, 27.3, 25.7, 25.6, 23.5, 23.0, 22.7, 17.9, 16.9, 16.6, 15.2; ESI-MS: 641.4 [(M+Na)+]. HRMS: m/z 617.4062 [M-H]- ([C36H57O8] = 617.4059).
Oleanolic acid 3-O-β-D-xylopyranoside (6c). Rf = 0.69 (5:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 5.45 (br s, 1H, H-C(12)), 4.84 (d, 1H, J=7.5, H-C(1')), 4.39 (dd, 1H, J=11.2, 4.9), 4.25-4.15 (m, 2H), 4.03 (t, 1H, J=7.8), 3.79 (t, 1H, J=10.4), 3.44 (br d, 1H, J=10.3, H-C(18)), 3.35 (dd, 1H, J=11.2, 3.8, H-C(3)), 1.31, 1.30, 1.02, 0.98, 0.95, 0.94, 0.85 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 180.4, 144.9, 122.5, 107.7, 88.6, 78.7, 75.6, 71.3, 67.2, 55.9, 48.1, 46.7, 46.5, 42.2, 42.0, 39.7, 39.6, 38.8, 37.0, 34.3, 33.3, 33.3, 33.2, 31.0, 28.3, 28.2, 26.8, 26.2, 23.8, 23.8, 23.7, 18.5, 17.4, 17.0, 15.5; ESI-MS: 611.5 [(M+Na)+]. HRMS: m/z 587.3953 [M-H]- ([C35H55O7] = 587.3953).
Oleanolic acid 3-O-β-D-galactopyranosyl-(1→4)-β-D-glucopyranoside (6e). Rf = 0.16 (4:1, CHCl3-MeOH); 1H-NMR (DMSO-d6) 5.16 (br s, 1H, H-C(12)), 4.23-4.21 (m, 2H, H-C(1'), H-C(1'')), 3.73-3.69 (br d, 1H, J=11.0), 3.61-3.44 (m, 5H), 3.33-3.25 (m, 5H), 3.04-3.01 (m, 2H, H-C(2'), H-C(3)), 2.73 (dd, 1H, J=12.8, 3.4, H-C(18)), 1.09 (s, 3H, CH3), 0.98 (s, 3H, CH3), 0.87 (s, 9H, 3×CH3), 0.75 (s, 3H, CH3), 0.71 (s, 3H, CH3); 13C-NMR (DMSO-d6) δ 178.8, 144.0, 121.7, 105.3, 104.1, 88.3, 81.3, 75.6, 75.2, 74.7, 73.7, 73.3, 70.7, 68.2, 60.8, 60.5, 55.2, 47.3, 45.9, 45.6, 41.5, 40.9, 38.8, 38.7, 38.3, 36.5, 33.5, 33.0, 32.6, 32.3, 30.6, 27.8, 27.4, 25.8, 25.8, 23.5, 23.1, 22.8, 18.0, 17.0, 16.7, 15.3; ESI-MS: 781.0 [(M+H)+], 798.0 [(M+NH4)+]. HRMS: m/z 779.4580 [M-H]- ([C42H67O13] = 779.4587).
Oleanolic acid 3-O-α-D-glucopyranosyl-(1→4)-β-D-glucopyranoside (6f). Rf = 0.13 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 5.78 (d, 1H, J=3.5, H-C(1'')), 5.50 (br s, 1H, H-C(12)), 5.32 (br d, 1H, J=11.5), 5.08 (dd, 1H, J=9.5, 5.6), 4.82 (d, 1H, J=7.7, H-C(1')), 4.63-4.50 (m, 3H), 4.43-4.31 (m, 2H), 4.24-4.04 (m, 5H), 3.28 (m, 2H, H-C(3), H-C(18)), 1.29, 1.25, 1.01, 0.96, 0.95, 0.90, 0.76 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 180.2, 144.9, 122.6, 106.8, 103.8, 89.6, 82.9, 77.9, 75.6, 75.4, 74.9, 74.4, 73.3, 71.6, 65.1, 62.5, 55.9, 48.0, 46.7, 46.5, 42.2, 42.0, 39.7, 39.4, 38.5, 36.9, 34.3, 33.3, 33.2, 33.2, 31.0, 28.3, 28.2, 26.5, 26.3, 23.8, 23.8, 23.7, 18.5, 17.4, 16.9, 15.4; ESI-MS: 779.2 [(M-H)-], 803.4 [(M+Na)+]. HRMS: m/z 779.4584 [M-H]- ([C42H67O13] = 779.4587).
Oleanolic acid 3-O-α-L-rhamnopyranoside (6g). Rf = 0.80 (5:1, CHCl3-MeOH); 1H-NMR (DMSO-d6) δ 12.05 (s, 1H, COOH), 5.16 (br s, 1H, H-C(12)), 4.70 (m, 2H, H-C(1'), OH), 4.58 (s, 1H, OH), 4.52 (d, 1H, J=5.8, OH), 3.62 (br s, 1H), 3.49 (m, 1H), 3.40 (m, 1H), 3.17 (m, 1H), 3.01 (dd, 1H, J=9.7, 3.0, H-C(3)), 2.74 (br d, 1H, J=10.8, H-C(18)), 1.10 (s, 3H, CH3), 1.09 (s, 3H, CH3), 0.87 (s, 9H, 3×CH3), 0.72 (s, 6H, 2×CH3); 13C-NMR (DMSO-d6) δ 178.7, 143.9, 121.6, 103.0, 87.6, 72.2, 70.8, 70.8, 68.6, 54.7, 47.1, 45.8, 45.5, 41.4, 40.9, 38.8, 38.6, 37.9, 36.4, 33.4, 32.9, 32.4, 32.2, 30.5, 28.0, 27.3, 25.7, 25.0, 23.5, 23.0, 22.7, 17.9, 17.8, 16.9, 16.5, 15.2; ESI-MS: 637.6 [(M+Cl)-]. HRMS: m/z 601.4104 [M-H]- ([C36H57O7] = 601.4110).
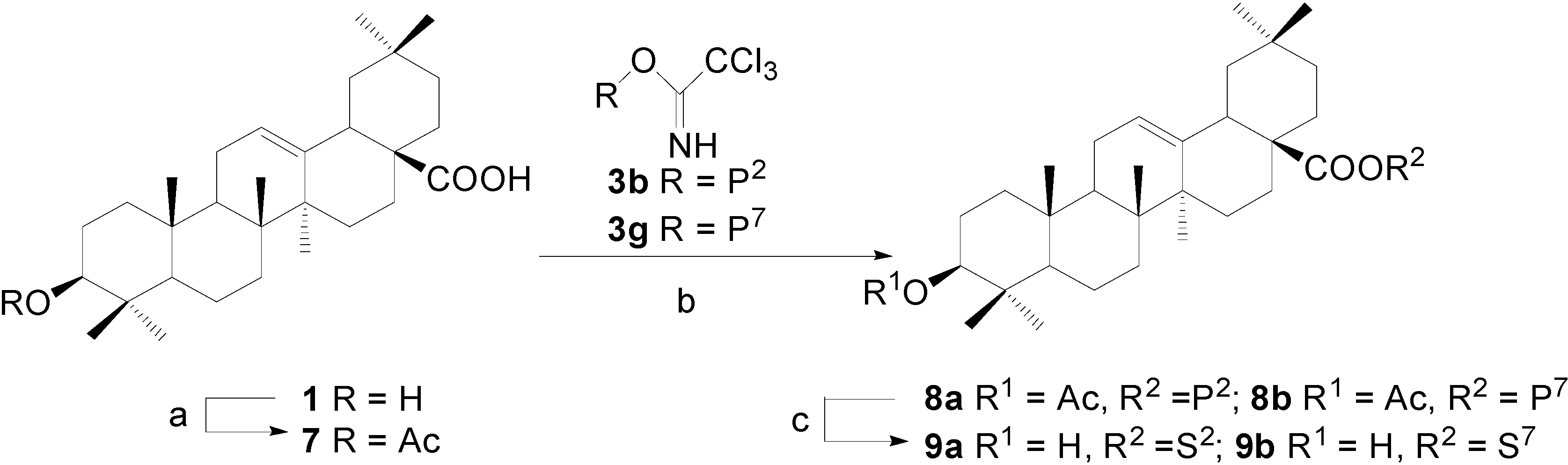
Synthesis of OA 28-glycosides 9a~9b, approach 1: β-D-galactopyranosyl oleanolate (9a)
Ac2O (18.9 mL) was added dropwise to a solution of OA (9.14 g, 20 mmol) in dry pyridine (38 mL) at 0°C under stirring. To the mixture was added DMAP (244 mg, 2 mmol) and the mixture was allowed to warm up to rt and stirred overnight. Water (15 mL) was added to quench the reaction. The mixture was then concentrated in vacuum and the residue was dissolved in CH2Cl2 (150 mL) and washed with 5% HCl, saturated NaHCO3 and brine in sequence. The solution was dried over Na2SO4. Recrystallization with MeOH-CH2Cl2 gave oleanolic acid 3-acetate (7) (7.66 g, 77%) as a white powder. Rf = 0.78 (3:1, petroleum ether-EtOAc); 1H-NMR (CDCl3) δ 5.27 (br s, 1H, H-C(12)), 4.49 (dd, 1H, J=8.2, 7.5, H-C(3)), 2.82 (br d, 1H, J=10.0, H-C(18)), 2.05 (s, 3H, Ac), 1.12, 0.94, 0.93, 0.90, 0.86, 0.85, 0.74 (s, 7×3H, CH3); ESI-MS: 521.8 [(M+Na)+].
A suspension of 7 (250 mg, 0.501 mmol), 3b (480 mg, 0.648 mmol) and powdered 4 Å molecular sieves (800 mg) in dry CH2Cl2 (8 mL) were stirred for 30 min at 0°C. TMSOTf (40 μL) was added and the mixture was stirred for 20 min before Et3N (0.15 mL) was added to quench the reaction. The mixture was then filtered and the filtrate was concentrated and purified by a silica gel column chromatography (7:1, petroleum ether-EtOAc) to afford 3-O-acetyloleanolic acid 2,3,4,6-tetra-O-benzoyl-β-D-galactopyranosyl ester (8a) (391 mg, 72%) as a white amorphous solid. Rf = 0.70 (4:1, petroleum ether-EtOAc); 1H-NMR (CDCl3) δ 8.10-7.21 (m, 20H, H-C(Ar)), 6.02 (d, 1H, J=3.3), 5.93 (m, 2H), 5.73 (m, 1H), 5.31 (br s, 1H, H-C(12)), 4.62 (dd, 1H, J=10.6, 6.1), 4.46-4.33 (m, 3H), 2.82 (dd, 1H, J=11.8, 2.1, H-C(18)), 2.04 (s, 3H, Ac), 0.97, 0.88, 0.85, 0.83, 0.82, 0.81, 0.49 (s, 7×3H, CH3); ESI-MS: 1099.8 [(M+Na)+].
8a (94 mg, 0.087 mmol) was dissolved in dry MeOH (8 ml), to which a freshly prepared solution of NaOMe in MeOH (1.0 M, 1.60 mL) was added. The mixture was stirred at rt for 12 h and then neutralized with Dowex H+ resin to pH 7 and filtered. The filtrate was concentrated and purified with a silica gel column chromatography (10:1, CHCl3-MeOH) to give β-D-galactopyranosyl oleanolate (9a) (17 mg, 32%) as a white powder. Rf = 0.43 (8:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.27 (d, 1H, J=8.0, H-C(1')), 5.44 (br s, 1H, H-C(12)), 4.69-4.64 (m, 2H), 4.48 (m, 1H), 4.40 (m, 1H), 4.21 (m, 2H), 3.43 (dd, 1H, J=9.5, 5.6, H-C(3)), 3.19 (br d, 1H, J=11.1, H-C(18)), 1.21, 1.21, 1.14, 1.02, 0.91, 0.88, 0.86 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 176.0, 143.7, 122.4, 95.8, 77.6, 77.3, 75.2, 71.0, 69.6, 61.4, 55.3, 47.7, 46.5, 45.7, 41.6, 41.3, 39.4, 38.9, 38.5, 36.9, 33.5, 32.7, 32.7, 32.0, 30.3, 28.3, 27.8, 27.6, 25.6, 23.4, 23.2, 22.8, 18.3, 17.0, 16.1, 15.2; ESI-MS: 641.4 [(M+Na)+]. HRMS: m/z 617.4058 [M-H]- ([C36H57O8] = 617.4059).
α-L-Rhamnopyranosyl oleanolate (9b)
This compound was prepared according to the same procedure described for 9a. Rf =0.60 (8:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.79 (s, 1H, J=1.4, H-C(1')), 5.43 (br s, 1H, H-C(12)), 4.58 (m, 1H), 4.51 (dd, 1H, J=8.6, 3.1), 4.41-4.36 (m, 2H), 3.44 (dd, 1H, J=9.4, 6.1, H-C(3)), 3.14 (br d, 1H, J=10.2, H-C(18)), 1.70 (d, 3H, J=5.4, H-C(6')), 1.23, 1.20, 1.04, 1.02, 0.90, 0.90, 0.86 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 175.9, 143.9, 122.6, 95.4, 78.1, 73.4, 72.9, 72.6, 71.6, 55.8, 48.0, 47.4, 46.0, 42.2, 42.1, 39.8, 39.4, 39.0, 37.4, 33.9, 33.3, 33.1, 33.0, 30.9, 28.8, 28.1, 28.0, 26.0, 23.9, 23.6, 23.3, 18.8, 18.8, 17.7, 16.6, 15.6; ESI-MS: 637.6 [(M+Cl)-]. HRMS: m/z 601.4105 [M-H]- ([C36H57O7] = 601.4110).
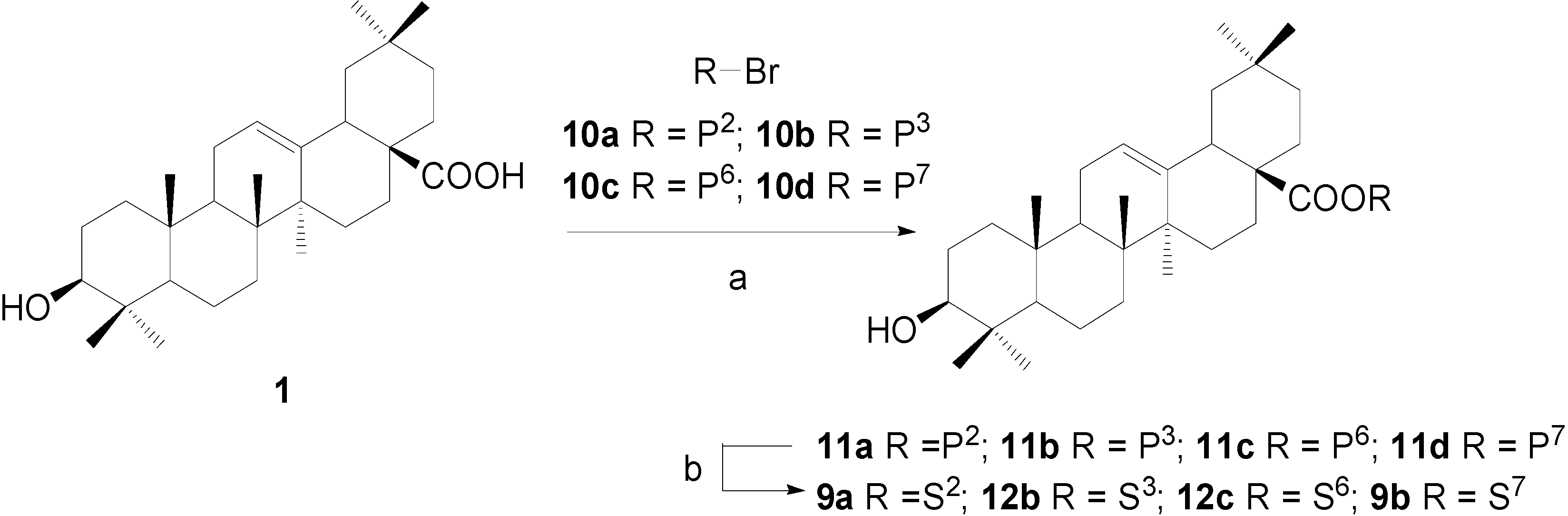
Synthesis of OA 28-glycosides 12a~12d, approach 2: β-D-xylopyranosyl oleanolate (12b)
OA (100 mg, 0.219 mmol), benzoylbromoglycoside 10b (138 mg, 0.263 mmol), and Bu4NBr (4 mg) were dissolved in CHCl3 (4.0 mL); K2CO3 (152 mg) was dissolved in water (1.5 mL). The two solutions were mixed together and stirred vigorously at 50°C for 2.5 h. The organic layer was separated and diluted with CHCl3 (10 mL) and washed by water (10 mL × 2) and concentrated to give a brown residue, which was subjected to a column chromatography (2:1, petroleum ether-EtOAc) to afford oleanolic acid 2,3,4-tri-O-benzoyl-β-D-xylopyranosyl ester (11b) (183 mg, 92%) as a white foam. Rf = 0.32 (3:1, petroleum-EtOAc); 1H-NMR (CDCl3) δ 8.01-7.32 (m, 15H, H-C(Ar)), 5.89 (m, 2H), 5.63 (dd, 1H, J=8.8, 7.4), 5.40 (td, 1H, J=8.8, 5.2), 5.28 (t, 1H, J=3.3, H-C(12)), 4.42 (dd, 1H, J=11.9, 5.1), 3.74 (dd, 1H, J=11.9, 8.9), 3.16 (dd, 1H, J=11.1, 4.2, H-C(3)), 2.81 (dd, 1H, J=13.4, 3.8, H-C(18)), 0.97, 0.95, 0.87, 0.85, 0.84, 0.76, 0.49 (s, 7×3H, CH3); ESI-MS: 918.8 [(M+NH4)+].
Compound 11b (190 mg, 0.211 mmol) was dissolved in dry CH2Cl2-MeOH (1:2, 6 mL) and a freshly prepared solution of NaOMe in MeOH (1.0 M, 0.50 mL) was added. The mixture was stirred at rt for 2 h and then neutralized with Dowex H+ resin to pH 7 and filtered. The filtrate was concentrated and purified with a silica gel column chromatography (12:1, CHCl3-MeOH) to give β-D-xylopyranosyl oleanolate (12b) (100 mg, 80%) as a white powder. Rf = 0.50 (10:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.24 (d, 1H, J=6.7, H-C(1')), 5.46 (br s, 1H, H-C(12)), 4.38 (dd, 1H, J=11.7, 4.4), 4.22 (m, 3H), 3.83 (m, 1H), 3.43 (m, 1H, H-C(3)), 3.25 (dd, 1H, J=11.8, 2.6, H-C(18)), 1.23 (s, 6H, 2×CH3), 1.10, 1.02, 0.92 (s, 3×3H, CH3), 0.90 (s, 6H, 2×CH3); 13C-NMR (pyridine-d5) δ 176.6, 144.1, 123.0, 96.3, 78.4, 78.1, 73.7, 70.9, 67.8, 55.8, 48.1, 47.2, 46.2, 42.2, 41.7, 39.9, 39.4, 38.9, 37.4, 34.0, 33.2, 33.1, 32.8, 30.8, 28.8, 28.3, 28.1, 26.1, 23.8, 23.6, 23.4, 18.8, 17.5, 16.6, 15.6; ESI-MS: 611.5 [(M+Na)+]. HRMS: m/z 587.3950 [M-H]- ([C35H55O7] = 587.3953).
β-D-Galactopyranosyl oleanolate (9a), α-D-glucopyranosyl-(1→4)-β-D-glucopyranosyl oleanolate (12c) and α-L-rhamnopyranosyl oleanolate (9b) were also prepared according to the same procedure described for 12b. The analytical data of 9a and 9b were identical to that of 9a and 9b prepared by approach 1.
α-D-Glucopyranosyl-(1→4)-β-D-glucopyranosyl oleanolate (12c). Rf = 0.23 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.23 (d, 1H, J=8.0, H-C(1')), 5.95 (d, 1H, J=3.2, H-C(1'')), 5.44 (br s, 1H, H-C(12)), 4.61-4.53 (m, 3H), 4.49 (m, 2H), 4.41-4.32 (m, 3H), 4.20-4.15 (m, 3H), 3.87 (br d, 1H, J=8.8), 3.43 (m, 1H, H-C(3)), 3.19 (br d, 1H, J=10.4, H-C(18)), 1.22 (s, 6H, 2×CH3), 1.10, 1.02 (s, 2×3H, CH3), 0.90 (s, 6H, 2×CH3), 0.88 (s, 3H, CH3); 13C-NMR (pyridine-d5) δ 176.4, 144.1, 123.2, 103.1, 95.5, 80.6, 78.3, 78.1, 77.6, 75.5, 75.4, 74.4, 73.6, 71.9, 62.7, 61.4, 55.8, 48.2, 47.0, 46.2, 42.2, 41.8, 39.9, 39.4, 39.0, 37.4, 34.0, 33.2, 33.2, 32.6, 30.8, 28.8, 28.3, 28.1, 26.1, 23.9, 23.7, 23.4, 18.8, 17.5, 16.6, 15.7; ESI-MS: 779.2 [(M-H)-]. HRMS: m/z 779.4585 [M-H]- ([C42H67O13] = 779.4587).
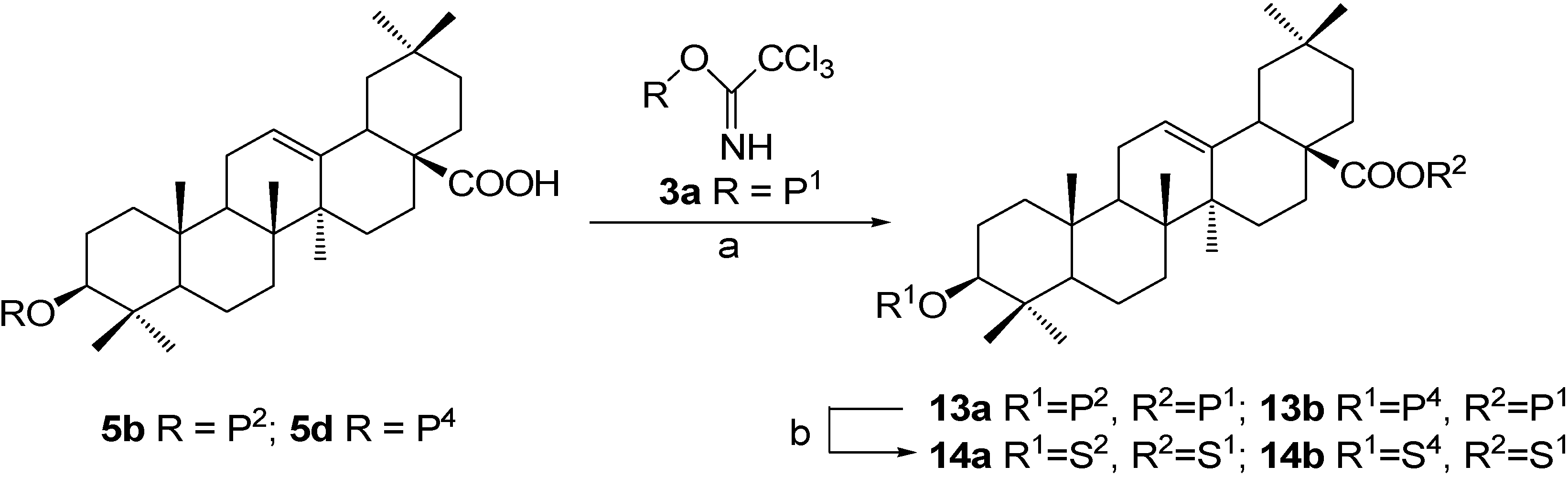
Synthesis of OA 3,28-diglycosides 14a~14b, approach 1: β-D-glucopyranosyl oleanolate 3-O-α-L-arabinopyranoside (14b).
A mixture of intermediate 5d (290 mg, 0.322 mmol), trichloroacetimidate 3a (286 mg, 0.386 mmol) and powdered 4 Å molecular sieves (1 g) in dry CH2Cl2 (8 mL) were stirred for 30 min at 0°C. A solution of TMSOTf in dry CH2Cl2 (5%, 0.50 mL) was added dropwise and the mixture was warmed to rt and stirred for 45 min before quenching the reaction with Et3N (0.10 mL). The mixture was then filtered and the filtrate was concentrated and purified by a silica gel column chromatography (3:1, petroleum ether-EtOAc) to afford 2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl oleanolate 3-O-(2,3,4-tri-O-benzoyl-α-L-arabinopyranoside) (13b) (438 mg, 92%) as a white powder. Rf = 0.22 (3:1, petroleum ether-EtOAc); 1H-NMR δ: (CDCl3) δ 8.08-7.25 (m, 35H, H-C(Ar)), 5.96 (dd, 1H, J=19.2, 9.7), 5.93 (d, 1H, J=8.3, H-C(1'')), 5.78-5.66 (m, 4H), 5.58 (dd, 1H, J=8.9, 3.4), 5.27 (br s, 1H, H-C(12)), 4.75 (d, 1H, J=6.5, H-C(1')), 4.56-4.43 (m, 2H), 4.34-4.23 (m, 2H), 3.86 (br d, 1H, J=11.6), 3.10 (dd, 1H, J=11.1, 4.8, H-C(3)), 2.77 (br d, 1H, J=10.9, H-C(18)), 0.93, 0.85, 0.82, 0.73, 0.73, 0.62, 0.41 (s, 7×3H, CH3); ESI-MS: 1501.9 [(M+Na)+].
Compound 13b (396 mg, 0.268 mmol) was dissolved in dry CH2Cl2-MeOH (1:2, 30 mL), to which a freshly prepared solution of NaOMe in MeOH (1.0 M, 1.20 ml) was added. The mixture was stirred at rt for 3 h and then neutralized with Dowex H+ resin to pH 7. The resin was then removed by filtration. Recrystallization with MeOH-Et2O furnished β-D-glucopyranosyl oleanolate 3-O-α-L-arabinopyranoside (14b) (133 mg, 66%) as a white powder. Rf = 0.46 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.33 (d, 1H, J=7.9, H-C(1'')), 5.42 (br s, 1H, H-C(12)), 4.75 (d, 1H, J=7.1, H-C(1')), 4.45-4.37 (m, 4H), 4.33-4.28 (m, 3H), 4.17-4.13 (m, 2H), 4.03 (m, 1H), 3.82 (br d, 1H, J=10.7), 3.34 (m, 1H, H-C(3)), 3.19 (dd, 1H, J=11.9, 2.0, H-C(18)), 1.26, 1.24, 1.09, 0.94, 0.90, 0.88, 0.86 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 176.4, 144.1, 122.9, 107.5, 95.8, 88.7, 79.3, 78.9, 74.6, 74.2, 72.9, 71.2, 69.5, 66.7, 62.3, 55.9, 48.1, 47.1, 46.3, 42.2, 41.8, 39.9, 39.6, 38.9, 37.1, 34.1, 33.2, 33.1, 32.6, 30.8, 28.3, 28.3, 26.6, 26.1, 23.9, 23.7, 23.5, 18.6, 17.5, 16.9, 15.6; ESI-MS: 785.2 [(M+Cl)-]. HRMS: m/z 750.4485 [M-H]- ([C41H65O12] = 750.4481).
β-D-Glucopyranosyl oleanolate 3-O-β-D-galactopyranoside (14a).
This substance was prepared according to the same procedure described for 14b. Rf = 0.24 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.36 (d, 1H, J=7.9, H-C(1'')), 5.45 (br s, 1H, H-C(12)), 4.88 (d, 1H, J=7.7, H-C(1')), 4.61 (d, 1H, J=3.2), 4.51-4.43 (m, 5H), 4.37-4.25 (m, 3H), 4.20-4.15 (m, 2H), 4.06 (m, 1H), 3.39 (dd, 1H, J=11.6, 3.6, H-C(3)), 3.22 (dd, 1H, J=13.2, 2.4, H-C(18)), 1.32, 1.29, 1.12, 0.98, 0.93, 0.90, 0.86 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 176.4, 144.2, 122.9, 107.5, 95.8, 88.8, 79.3, 78.9, 76.8, 75.5, 74.2, 73.2, 71.2, 70.3, 62.5, 62.3, 55.9, 48.1, 47.0, 46.3, 42.2, 41.8, 40.0, 39.6, 38.8, 37.0, 34.1, 33.2, 33.2, 32.6, 30.8, 28.3, 28.3, 26.7, 26.2, 23.8, 23.7, 23.5, 18.6, 17.5, 17.0, 15.6; ESI-MS: 781.0 [(M+H)+], 1561.1 [(2M+H)+]. HRMS: m/z 779.4580 [M-H]- ([C42H67O13] = 779.4587).
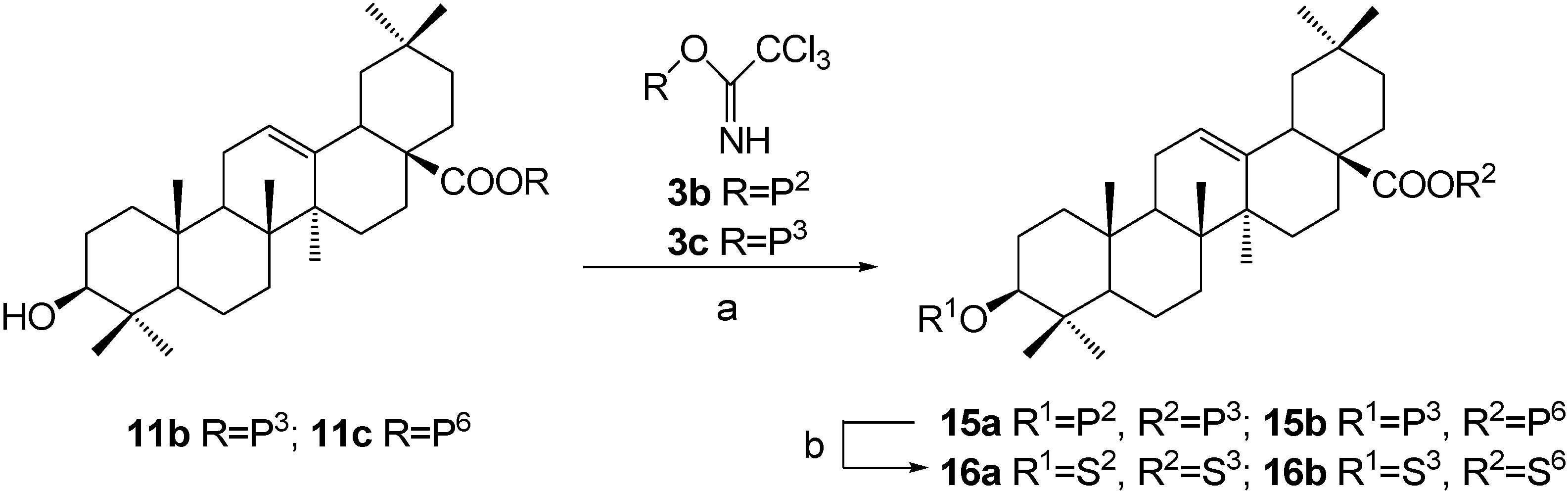
Synthesis of OA 3,28-diglycosides 16a~16b, approach 2: β-D-xylopyranosyl oleanolate 3-O-β-D-galactopyranoside (16a).
A mixture of intermediate 11b (600 mg, 0.666 mmol), trichloroacetimidate 3b (570 mg, 0.765 mmol) and powdered 4 Å molecular sieves (600 mg) in dry CH2Cl2 (9 mL) were stirred for 30 min at rt. A solution of TMSOTf in dry CH2Cl2 (5%, 128 μL) was added dropwise and the mixture was stirred for 30 min before Et3N (0.40 ml) was added to quench the reaction. The mixture was then filtered and the filtrate was concentrated and purified by a silica gel column chromatography (3:1, petroleum ether-EtOAc) to afford 2,3,4-tri-O-benzoyl-β-D-xylopyranosyl oleanolate 3-O-(2,3,4,6-tetra-O-benzoyl-β-D-galactocopyranoside) (15a) (920 mg, 93.4%) as a white powder. Rf = 0.23 (3:1, petroleum ether-EtOAc); 1H-NMR (CDCl3) δ 8.12-7.22 (m, 35H, H-C(Ar)), 5.95 (s, 1H, H-C(4')), 5.90 (t, 1H, J=8.8, H-C(2'')), 5.87 (d, 1H, J=7.4, H-C(1'')), 5.83 (t, 1H, J=9.2, H-C(2')), 5.64-5.59 (m, 2H, H-C(3'), H-C(3'')), 5.40 (m, 1H, H-C(4'')), 5.30 (br s, 1H, H-C(12)), 4.81 (d, 1H, J=7.9, H-C(1')), 4.65 (dd, 1H, J=11.1, 7.7, H-C(6')-1), 4.44-4.41 (m, 2H, H-C(6')-2, H-C(5'')-1), 4.30 (m, 1H, H-C(5')), 3.72 (dd, 1H, J=14.7, 6.8, H-C(5'')-2), 3.09 (dd, 1H, J=11.7, 4.3, H-C(3)), 2.81 (br d, 1H, J=10.8, H-C(18)), 0.94, 0.88, 0.87, 0.79, 0.67, 0.66, 0.44 (s, 7×3H, CH3); ESI-MS: 1478.0 [(M-H)-], 1501.9 [(M+Na)+].
Compound 15a (340 mg, 0.230 mmol) was dissolved in dry CH2Cl2-MeOH (1:2, 36 mL), to which a freshly prepared solution of NaOMe in MeOH (1.0 M, 1.60 mL) was added. The solution was stirred at rt for 12 h and then neutralized with Dowex H+ resin to pH 7. The resin was then removed by filtration. Recrystallization with MeOH-Et2O furnished β-D-xylopyranosyl oleanolate 3-O-β-D-galactopyranoside (16a) (132 mg, 76.5%) as a white powder. Rf = 0.41 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.22 (d, 1H, J=6.4, H-C(1'')), 5.43 (br s, 1H, H-C(12)), 4.86 (d, 1H, J=7.6, H-C(1')), 4.58 (s, 1H, H-C(4')), 4.48-4.42 (m, 3H, H-C(2'), H-C(6')-1, H-C(3'')), 4.36 (dd, 1H, J=11.6, 4.3, H-C(5'')-1), 4.22 (m, 1H, H-C(4'')), 4.21-4.15 (m, 3H, H-C(3'), H-C(6')-2, H-C(2'')), 4.12 (m, 1H, H-C(5')), 3.80 (m, 1H, H-C(5'')-2), 3.36 (dd, 1H, J=18.7, 6.0, H-C(3)), 3.23 (br d, 1H, J=10.4, H-C(18)), 1.29, 1.25, 1.06, 0.96, 0.92, 0.90, 0.83 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 176.6, 144.1, 122.9, 107.6, 96.3, 88.7, 78.4, 76.9, 75.5, 73.7, 73.2, 70.9, 70.3, 67.8, 62.5, 55.8, 48.0, 47.1, 46.2, 42.1, 41.7, 39.9, 39.5, 38.7, 36.9, 34.0, 33.1, 33.1, 32.7, 30.8, 28.2, 28.2, 26.7, 26.1, 23.8, 23.6, 23.3, 18.5, 17.5, 17.0, 15.5; ESI-MS: 773.4 [(M+Na)+]. HRMS: m/z 750.4483 [M-H]- ([C41H65O12] = 750.4481).
α-D-Glucopyranosyl-(1→4)-β-D-glucopyranosyl oleanolate 3-O-β-D-xylopyranoside (16b).
This compound was prepared according to the same procedure described for 16a. Rf = 0.19 (4:1, CHCl3-MeOH); 1H-NMR (pyridine-d5) δ 6.22 (d, 1H, J=8.2, H-C(1'')), 5.94 (d, 1H, J=3.5, H-C(1''')), 5.42 (br s, 1H, H-C(12)), 4.82 (d, 1H, J=7.5, H-C(1')), 4.58-4.53 (m, 3H), 4.45 (m, 2H), 4.37 (m, 3H), 4.33 (t, 1H, J=5.8), 4.22 (m, 1H), 4.17-4.14 (m, 4H), 4.01 (t, 1H, J=8.2), 3.87 (br d, 1H, J=8.2), 3.77 (t, 1H, J=11.5), 3.34 (dd, 1H, J=11.7, 4.2, H-C(3)), 3.17 (dd, 1H, J=10.9, 3.9, H-C(18)), 1.29, 1.25, 1.07, 0.98, 0.90, 0.88, 0.86 (s, 7×3H, CH3); 13C-NMR (pyridine-d5) δ 176.4, 144.1, 123.0, 107.8, 103.1, 95.5, 88.7, 80.6, 78.7, 78.3, 77.6, 75.6, 75.4, 75.4, 74.5, 73.6, 71.9, 71.3, 67.2, 62.8, 61.4, 55.9, 48.1, 47.0, 46.2, 42.2, 41.8, 40.0, 39.6, 38.8, 37.1, 34.0, 33.2, 33.2, 32.6, 30.8, 28.2, 28.2, 26.8, 26.1, 23.9, 23.7, 23.4, 18.5, 17.5, 17.0, 15.6; ESI-MS: 935.5 [(M+Na)+]. HRMS: m/z 911.5011 [M-H]- ([C47H75O17] = 911.5011).








{kind=link}
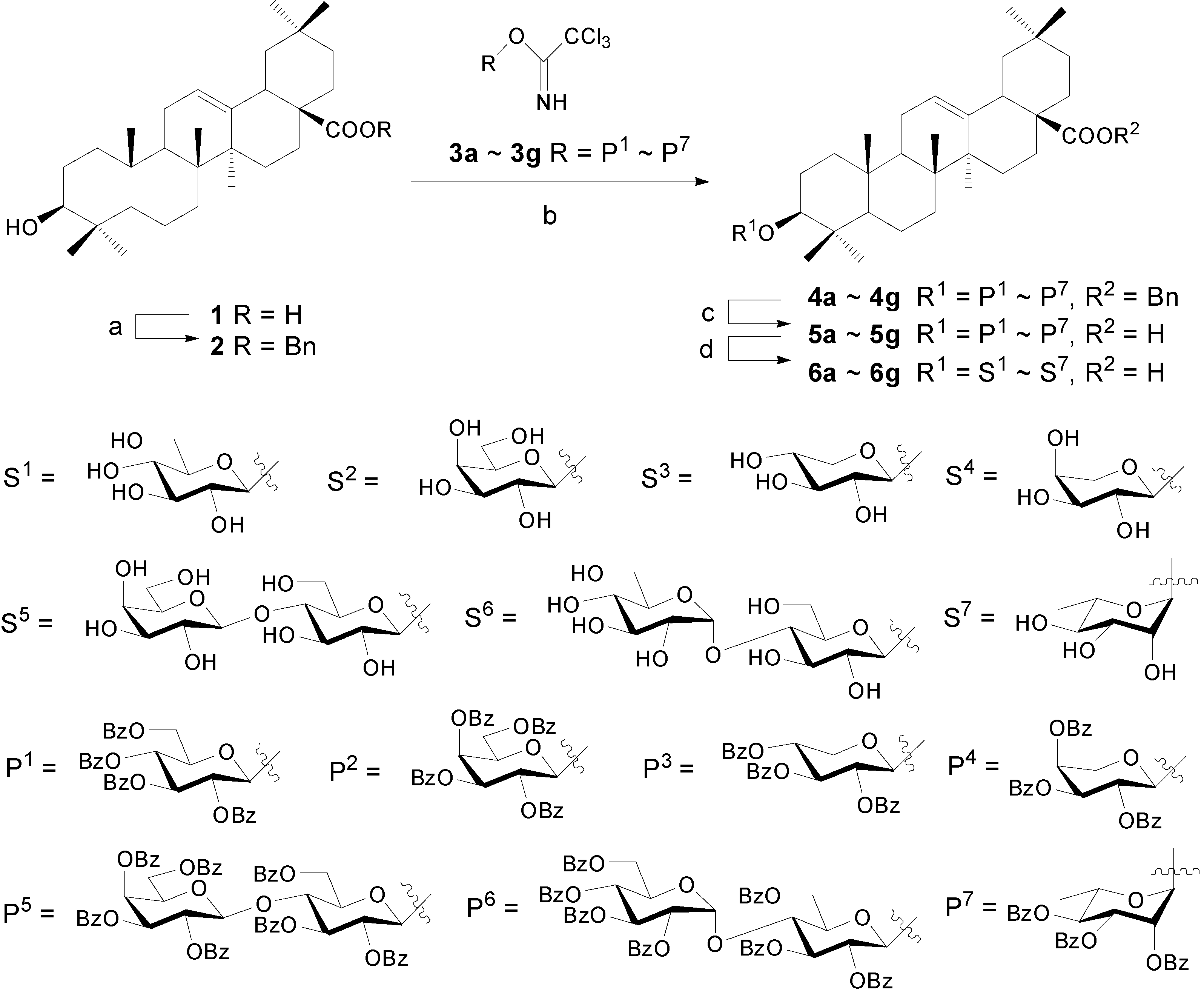{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}