New Acetylenic Norlignan Compounds from Rhizomes of Curculigo crassifolia

Abstract
:Introduction
Results and Discussion

{kind=link}
| NO. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 4.38 d (6.28) | 4.47 d (3.76) | 4.08 d (8.20) | 4.09 d (3.40) |
| 2 | 4.14 m | 4.14 m | 3.82 m | 3.75 m |
| 3 | 2.70 dd (17.12, 4.76) | 2.56 dd (13.84, 5.28) | 2.46 dd (16.65, 4.30) | 2.46 dd (16.65, 4.30) |
| 2.30 dd (17.12, 5.20) | 2.30 dd (13.84, 4.56) | 2.20 dd (16.65, 5.95) | 2.20 dd (16.65, 5.95) | |
| 2' | 6.89 d (1.44) | 6.89 d (1.44) | 6.87 d (1.50) | 6.87 d (1.50) |
| 5' | 6.81 d (8.16) | 6.81 d (8.16) | 6.80 d (8.00) | 6.80 d (8.00) |
| 6' | 6.75 dd (8.16, 1.44) | 6.75 dd (8.16, 1.44) | 6.75 dd (8.00, 1.50) | 6.75 dd (8.00, 1.50) |
| 2'' | 6.87 d (1.58) | 6.87 d (1.58) | 6.82 d (2.00) | 6.82 d (2.00) |
| 5'' | 6.71 d (8.12) | 6.71 d (8.12) | 6.71 d (8.08) | 6.71 d (8.08) |
| 6'' | 6.80 dd (8.12, 1.58) | 6.80 dd (8.12, 1.58) | 6.78 dd (8.08, 2.00) | 6.78 dd (8.08, 2.00) |
| OMe | 3.25 s | 3.37 s | 3.18 s | 3.18 s |
| Glc. | ||||
| 1 | 4.63 d (7.56) | 4.60 d (7.80) | ||
| 2 | 3.30- 3.42 m | 3.30- 3.42 m | ||
| 3 | 3.30- 3.42 m | 3.30- 3.42 m | ||
| 4 | 3.30- 3.42 m | 3.30- 3.42 m | ||
| 5 | 3.30- 3.42 m | 3.30- 3.42 m | ||
| 6 | 3.89 dd (11.84, 2.00) | 3.89 dd (11.84, 2.00) | ||
| 3.70 dd (11.84, 5.32) | 3.70 dd (11.84, 5.32) |
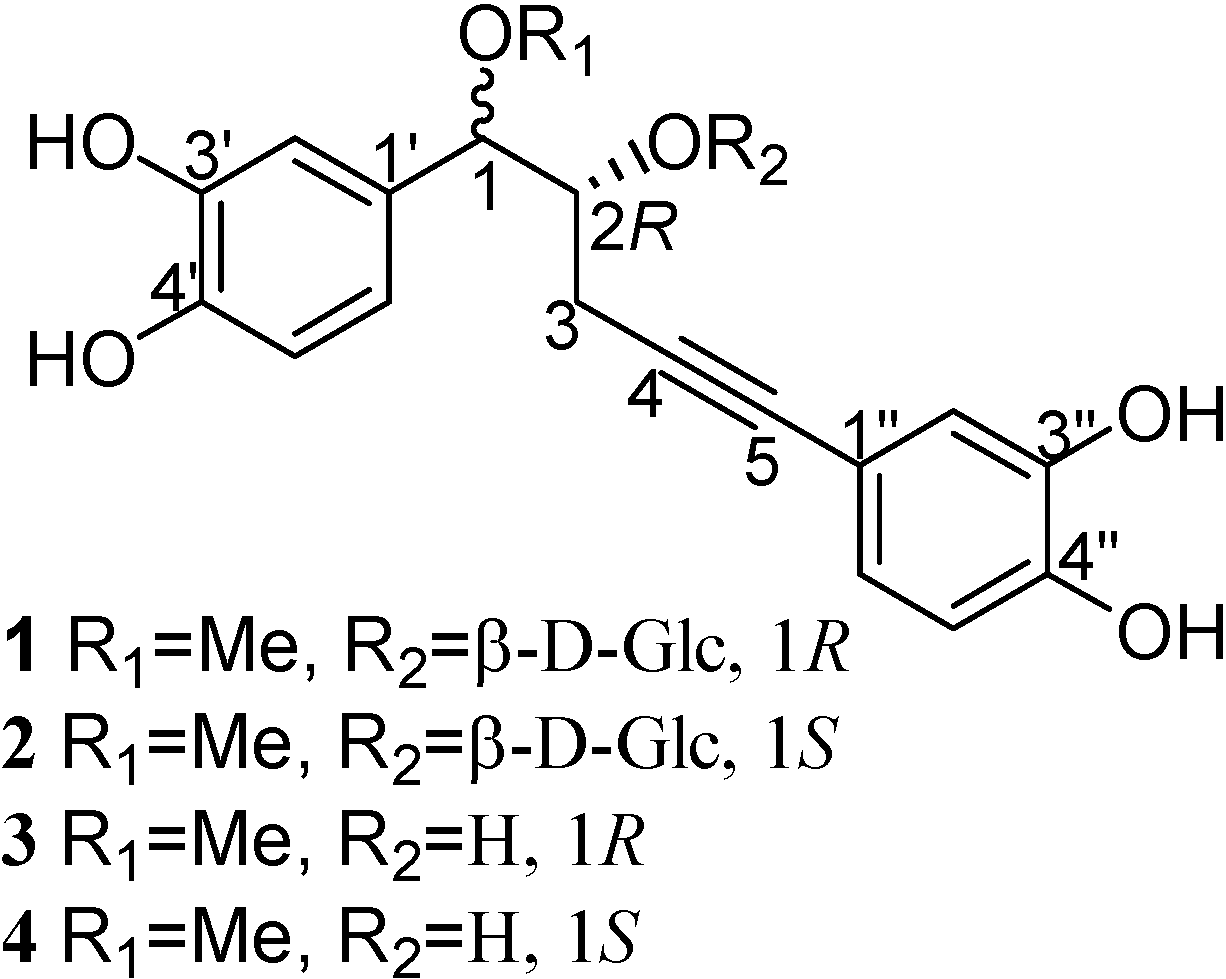
 +26.50º (c 0.16, MeOH), was obtained as a white amorphous powder and assigned a molecular formula of C24H28O11 on the basis of the HRFAB-MS (-) (m/z 491.1565 [M-1]-, calcd. 491.1553). The IR absorption at 3441 cm-1 indicated the presence of hydroxyl groups. The 1H-NMR spectrum displayed signals for six aromatic protons in two ABX systems, and seven sugar protons, in addition to signals for four aliphatic protons at δ 4.38 (d, H-1), 4.14 (m, H-2), 2.30 (dd, H-3), and 2.70 (dd, H-3). Both sets of ABX systems, one at 6.89 (d, J = 1.44 Hz, H-2'), 6.81 (d, J = 8.16 Hz, H-5'), and 6.75 (dd, J = 8.16, 1.44 Hz, H-6') and the other at 6.87 (d, J = 1.58 Hz, H-2''), 6.71 (d, J = 8.12 Hz, H-5''), and 6.80 (dd, J = 8.12, 1.58 Hz, H-6''), were consistent with two catechol-like moieties, with the latter being conjugated with a acetylene function (δ 84.4, 83.7). Analysis of the signals of seven sugar protons suggested a β-D-glucosyl unit with the anomeric proton at δ 4.63 (d, J = 7.56 Hz). These assignments were made by analyzing the H-H COSY spectrum, incorporating HMQC data. The placement of 1-O-methyl and 2-O-β-D-Glc was made from the observation of the three-bond coupling of H-1 to C-1 of the methyl group, anomeric proton to C-2, and H-2 to the anomeric carbon in the HMBC spectrum. The two remaining quaternary carbon signals (δ 84.4, 83.7) belong to the acetylenic bond. The HMBC spectrum also revealed couplings of H-2 and H-3 to C-4, H-2' and H-6' to C-5. Taking all these chemical shifts and their coupling relationships into consideration, the structure sequence of PhCH(OR1)CH(OR2)CH2C≡CPh for 1 was arrived at, allowing the attachment of a methoxyl group at C-1 position and the β-D-Glc moiety at the C-2 position (Figure 1).
+26.50º (c 0.16, MeOH), was obtained as a white amorphous powder and assigned a molecular formula of C24H28O11 on the basis of the HRFAB-MS (-) (m/z 491.1565 [M-1]-, calcd. 491.1553). The IR absorption at 3441 cm-1 indicated the presence of hydroxyl groups. The 1H-NMR spectrum displayed signals for six aromatic protons in two ABX systems, and seven sugar protons, in addition to signals for four aliphatic protons at δ 4.38 (d, H-1), 4.14 (m, H-2), 2.30 (dd, H-3), and 2.70 (dd, H-3). Both sets of ABX systems, one at 6.89 (d, J = 1.44 Hz, H-2'), 6.81 (d, J = 8.16 Hz, H-5'), and 6.75 (dd, J = 8.16, 1.44 Hz, H-6') and the other at 6.87 (d, J = 1.58 Hz, H-2''), 6.71 (d, J = 8.12 Hz, H-5''), and 6.80 (dd, J = 8.12, 1.58 Hz, H-6''), were consistent with two catechol-like moieties, with the latter being conjugated with a acetylene function (δ 84.4, 83.7). Analysis of the signals of seven sugar protons suggested a β-D-glucosyl unit with the anomeric proton at δ 4.63 (d, J = 7.56 Hz). These assignments were made by analyzing the H-H COSY spectrum, incorporating HMQC data. The placement of 1-O-methyl and 2-O-β-D-Glc was made from the observation of the three-bond coupling of H-1 to C-1 of the methyl group, anomeric proton to C-2, and H-2 to the anomeric carbon in the HMBC spectrum. The two remaining quaternary carbon signals (δ 84.4, 83.7) belong to the acetylenic bond. The HMBC spectrum also revealed couplings of H-2 and H-3 to C-4, H-2' and H-6' to C-5. Taking all these chemical shifts and their coupling relationships into consideration, the structure sequence of PhCH(OR1)CH(OR2)CH2C≡CPh for 1 was arrived at, allowing the attachment of a methoxyl group at C-1 position and the β-D-Glc moiety at the C-2 position (Figure 1).
| NO. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 85.8 d | 85.6 d | 86.4 d | 87.0 d |
| 2 | 79.5 d | 79.4 d | 73.9 d | 74.5 d |
| 3 | 22.4 t | 22.3 t | 24.9 t | 24.5 t |
| 4 | 84.4 s | 84.7 s | 85.6 s | 85.2 s |
| 5 | 83.7 s | 83.6 s | 82.7 s | 82.8 s |
| 1' | 130.4 s | 130.2 s | 131.3 s | 131.3 s |
| 2' | 116.0 d | 116.0 d | 115.8 da | 115.8 da |
| 3' | 145.8 s | 145.8 s | 145.6 s | 145.6 s |
| 4' | 146.3 s | 146.3 s | 145.9 s | 145.9 s |
| 5' | 116.0 d | 116.0 d | 115.9 da | 115.9 da |
| 6' | 120.8 d | 120.6 d | 120.7 d | 120.2 d |
| 1'' | 116.2 s | 116.2 s | 116.3 s | 116.3 s |
| 2'' | 119.4 d | 119.4 d | 119.2 d | 119.2 d |
| 3'' | 146.1 s | 146.1 s | 145.8 s | 145.8 s |
| 4'' | 146.7 s | 146.7 s | 146.3 s | 146.3 s |
| 5'' | 116.2 d | 116.2 d | 116.1 d | 116.1 d |
| 6'' | 124.9 d | 124.9 d | 124.6 d | 124.6 d |
| OMe | 57.1 q | 57.3 q | 56.8 q | 56.8 q |
| Glc. | ||||
| 1 | 102.4 d | 102.7 d | ||
| 2 | 74.7 d | 74.7 d | ||
| 3 | 77.6 d | 77.6 d | ||
| 4 | 71.3 d | 71.3 d | ||
| 5 | 77.8 d | 77.8 d | ||
| 6 | 62.6 t | 62.6 t |
Experimental
General
Plant material
Extraction and isolation
= 0º (c 0.12, MeOH); UV (MeOH): λmax (lg ε): 205 (4.56), 256 (4.08), 289 (3.77) nm; IR νmax: 3441, 2924, 1629, 1517, 1443, 1283, 1179, 1111, 815, 583cm-1; 1H-NMR see Table 1, 13C-NMR see Table 2; FAB-MS m/z: 329 [M-H]-; HR-FAB- MS m/z: [M-H]-329.1037 (calcd. for C18H17O6, 329.1025). Fraction 5 (210 g) was refractionated by Sephadex LH-20 (EtOH-H2O, 0:1-1:0; 2000 mL each eluent) to yield 12 crude fractions (5-1 to 5-12). Fraction 5-7 (4.34 g) was purified by Sephadex LH-20 (EtOH-H2O, 0:1-1:0; 700 ml each eluent) to yield 6 fractions (5-7-1 to 5-7-6). Fraction 5-7-4 (612 mg) was repeatedly purified on Sephadex LH-20 (EtOH) to afford a mixture of (1R, 2R)-1-O-methylnyasicoside (1) and (1S, 2R)-1-O-methylnyasicoside (2) (212 mg) and pure 1 (18 mg). White amorphous powder; : +12.37º (c 0.18, MeOH); UV (MeOH): λmax (lg ε): 205 (4.34), 255 (3.96), 289 (3.65) nm. IR νmax: 3441, 2926, 2045, 1546, 1473, 1179cm-1; 1H-NMR see Table 1; 13C-NMR see Table 2; FAB-MS m/z: 491 [M-H]-; HR-FAB-MS m/z: [M-H]- 491.1565 (calcd. for C24H27O11, 491.1553).Acknowledgements
References
- Institutum Botanicum Kunmingense, Academiae Sinicae. In Flora Yunnanica; Wu, C.Y. (Ed.) Science Press: Beijing; Vol. 6, p. 819. (in Chinese)
- Li, N.; Wang, K.J.; Chen, J.J.; Zhou, J. Two novel glucosyl-fused compounds from Curculigo crassifolia (Hypoxidaceae). Tetrahedron Lett. 2005, 46, 6445–6447. [Google Scholar] [CrossRef]
- Li, N.; Chen, J.J.; Zhou, J. Two New Phenolic Glycosides from Rhizomes of Curculigo crassifolia. Z. Naturforsch. B. 2006, 61b, 611–614. [Google Scholar]
- Wang, K.J.; Li, N. Antioxidant Phenolic Compounds from Rhizomes of Curculigo crassifolia. Arch. Pharm. Res. 2007, 30, 8–12. [Google Scholar] [CrossRef]
- Chang, W.L.; Su, M.J.; Lee, S.S. Bioactive norlignan glucosides from Curculigo capitulata. J. Nat. Prod. 1997, 60, 76–80. [Google Scholar]
- Chang, W.L.; Chen, C.H.; Lee, S.S. Three novel constituents from Curculigo capitulata and revision of C-2 stereochemistry in nyasicoside. J. Nat. Prod. 1999, 62, 734–739. [Google Scholar]
- Sample Availability: Available from authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, K.-j.; Li, N.; Wang, H. New Acetylenic Norlignan Compounds from Rhizomes of Curculigo crassifolia. Molecules 2008, 13, 1696-1701. https://doi.org/10.3390/molecules13081696
Wang K-j, Li N, Wang H. New Acetylenic Norlignan Compounds from Rhizomes of Curculigo crassifolia. Molecules. 2008; 13(8):1696-1701. https://doi.org/10.3390/molecules13081696
Chicago/Turabian StyleWang, Kai-jin, Ning Li, and Hu Wang. 2008. "New Acetylenic Norlignan Compounds from Rhizomes of Curculigo crassifolia" Molecules 13, no. 8: 1696-1701. https://doi.org/10.3390/molecules13081696
APA StyleWang, K. -j., Li, N., & Wang, H. (2008). New Acetylenic Norlignan Compounds from Rhizomes of Curculigo crassifolia. Molecules, 13(8), 1696-1701. https://doi.org/10.3390/molecules13081696