Amifostine Protection Against Mitomycin-induced Chromosomal Breakage in Fanconi Anaemia Lymphocytes

Abstract
:Introduction
Results and Discussion

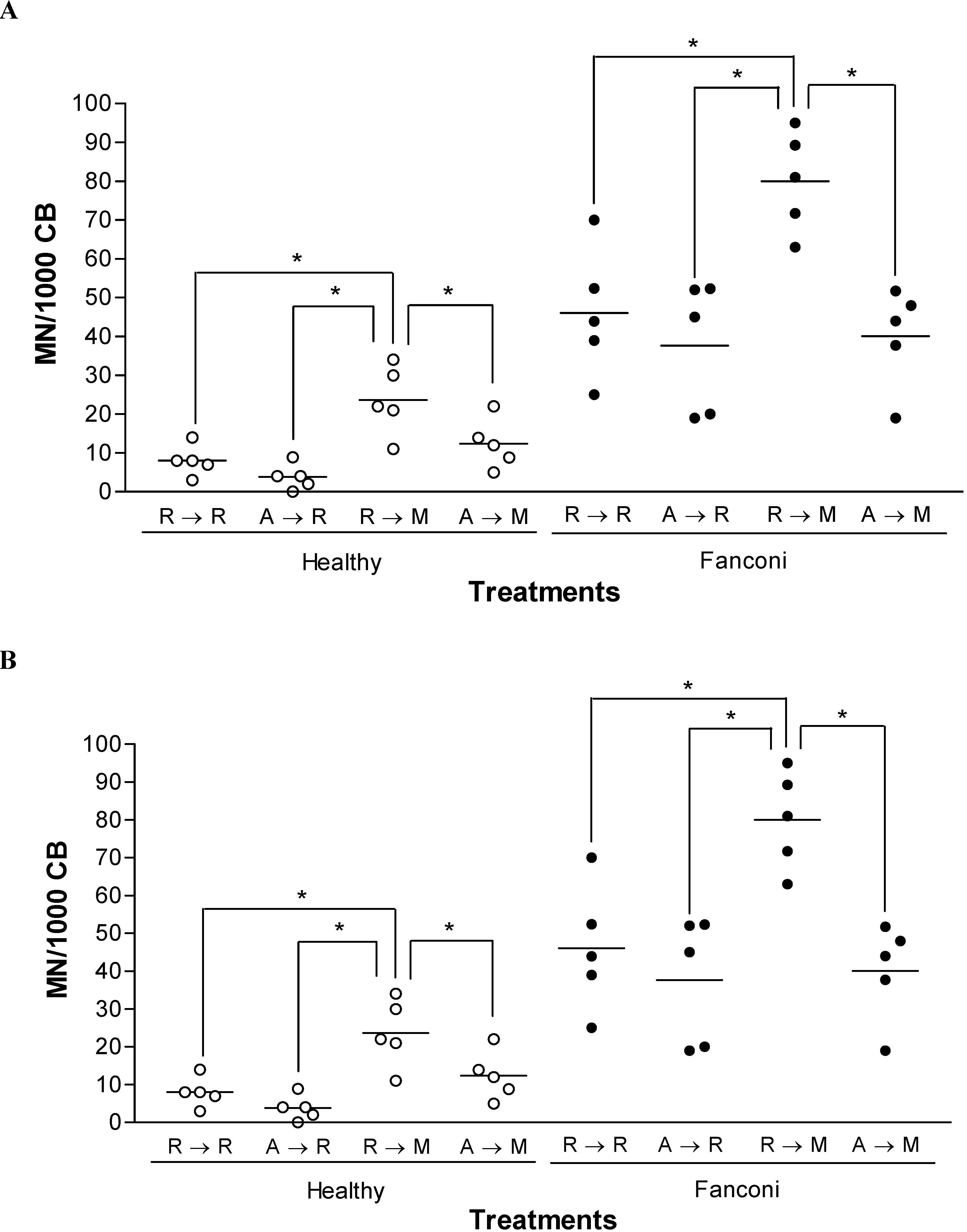{kind=link}
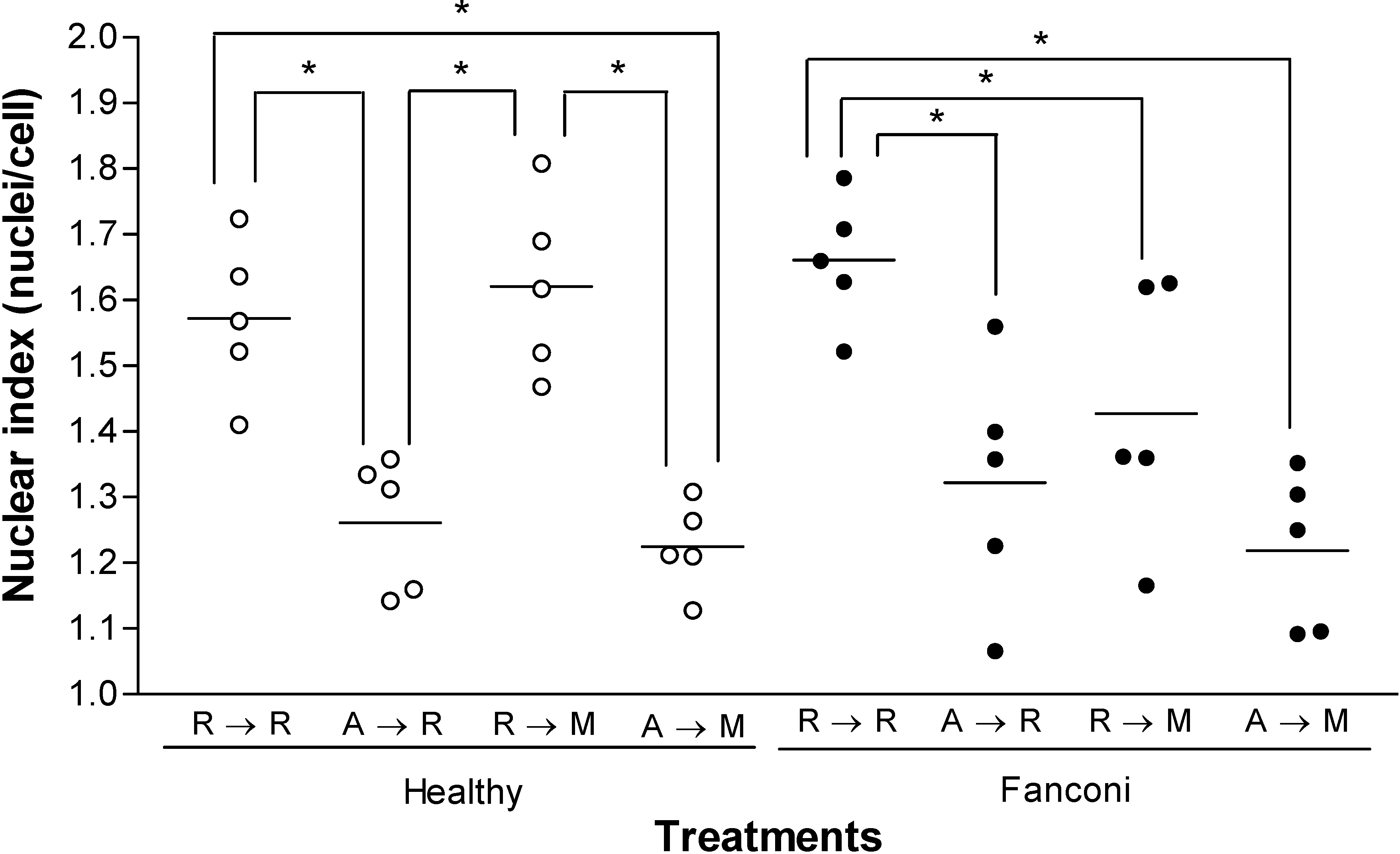{kind=link}
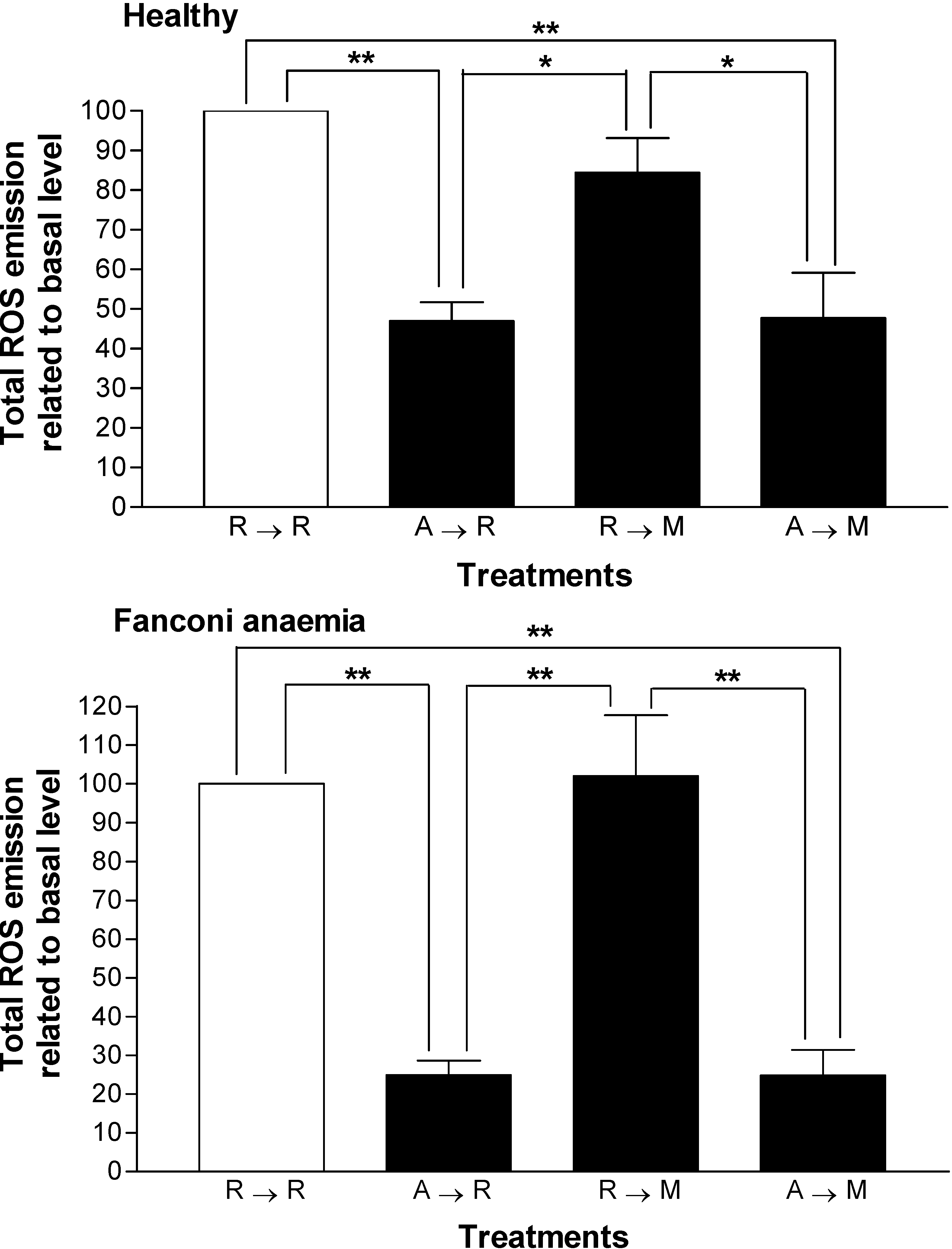{kind=link}
| Control probands | FA probands | |||||||
|---|---|---|---|---|---|---|---|---|
| Code | Age | Sex | Code | Age | Sex | Chromosomal breakage | OXM | |
| Spontaneous | MMC | |||||||
| FMG | 20 | M | ACS | 8 | M | 0.90 | 10.32 | - |
| FSC | 18 | F | CCF | 12 | F | 0.50 | 1.16 | + |
| LES | 15 | M | CBS | 24 | F | 2.16 | 12.91 | - |
| LGBC | 14 | M | EMM | 17 | M | 1.20 | 6.12 | + |
| SSS | 25 | F | RGS | 8 | F | 0.84 | 7.00 | - |
| TMBC | 17 | F | VBM | 7 | M | 0.80 | 4.80 | - |
| VALA | 13 | M | 0.92 | 2.56 | + | |||



Experimental
Subject eligibility
Sample collection and isolation of peripheral blood mononuclear cells (PBMC)
Chemicals and culture medium
Alkaline phosphatase activity
Micronucleus assay
Chemiluminescence analysis
Statistical analysis
Acknowledgements
References
- Alter, B.P. Cancer in Fanconi anemia, 1927-2001. Cancer 2003, 97, 425–440. [Google Scholar] [CrossRef]
- Papadopoulo, D.; Moustacchi, E. L’anémie de Fanconi: gènes et fonction(s) revisités. Med. Sci. 2005, 21, 730–736. [Google Scholar]
- Levitus, M.; Joenje, H.; de Winter, J.P. The Fanconi anemia pathway of genomic maintenance. Cell Oncol. 2006, 28, 3–29. [Google Scholar]
- Liu, J.M.; Buchwald, M.; Walsh, C.E.; Young, N.S. Fanconi anemia and novel strategies for therapy. Blood 1994, 84, 3995–4007. [Google Scholar]
- Velazquez, I.; Alter, B.P. Androgens and liver tumors: Fanconi’s anemia and non-Fanconi’s conditions. Am. J. Hematol. 2004, 77, 257–267. [Google Scholar] [CrossRef]
- Gluckman, E.; Devergie, A.; Dutreix, J. Radiosensitivity in Fanconi anaemia: application to the conditioning regimen for bone marrow transplantation. Br. J. Haematol. 1983, 54, 431–440. [Google Scholar] [CrossRef]
- Zanis-Neto, J.; Flowers, M.E.; Medeiros, C.R.; Bitencourt, M.A.; Bonfim, C.M.; Setúbal, D.C.; Funke, V.; Sanders, J.; Deeg, H.J.; Kiem, H.P.; Martin, P.; Leisenring, W.; Storb, R.; Pasquini, R. Low-dose cyclophosphamide conditioning for haematopoietic cell transplantation from HLA-matched related donors in patients with Fanconi anaemia. Br. J. Haematol. 2005, 130, 99–106. [Google Scholar] [CrossRef]
- Rosenberg, P.S.; Greene, M.H.; Alter, B.P. Cancer incidence in persons with Fanconi’s anemia. Blood 2003, 101, 822–826. [Google Scholar] [CrossRef]
- Nordenson, I. Effect of superoxide dismutase and catalase on spontaneously occurring chromosome breaks in patients with Fanconi’s anemia. Hereditas 1977, 86, 147–150. [Google Scholar] [CrossRef]
- Joenje, H.; Arwert, F.; Eriksson, A.W.; de Koning, H.; Oostra, A.B. Oxygen-dependence of chromosomal aberrations in Fanconi’s anaemia. Nature 1981, 290, 142–143. [Google Scholar] [CrossRef]
- Korkina, L.G.; Deeva, I.B.; Iaccarino, M.; Oral, R.; Warnau, M.; Pagano, G. Redox dependent toxicity of diepoxybutane and mitomycin C in sea urchin embryogenesis. Carcinogenesis 2000, 21, 213–220. [Google Scholar] [CrossRef]
- Raj, A.S.; Heddle, J.A. The effect of superoxide dismutase, catalase and L-cysteine on spontaneous and on mitomycin C induced chromosomal breakage in Fanconi’s anemia and normal fibroblasts as measured by the micronucleus method. Mutat. Res. 1980, 78, 59–66. [Google Scholar] [CrossRef]
- Ruppitsch, W.; Meisslitzer, C.; Hirsch-Kauffmann, M.; Schweiger, M. Overexpression of thioredoxin in Fanconi anemia fibroblasts prevent the cytotoxic and DNA damaging effect of mitomycin C and diepoxybutane. FEBS Lett. 1998, 422, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Schuchter, L.M.; Hensley, M.L.; Meropol, L.J.; Winer, E.P. American Society of Clinical Oncology, Chemotherapy and Radiotherapy Expert Panel 2002 update of recommendations for the use of chemotherapy and radiotherapy protectants: clinical practice guidelines of the American Society of Clinical Oncology. J. Clin. Oncol. 2002, 20, 2895–2903. [Google Scholar] [CrossRef]
- Yuhas, J.M.; Storer, J.B. Differential chemoprotection of normal and malignant tissues. J. Natl. Cancer Inst. 1969, 42, 331–335. [Google Scholar]
- Calabro-Jones, P.M.; Fahey, R.C.; Smoluk, G.D.; Ward, J.F. Alkaline phosphatase promotes radioprotection and accumulation of WR-1065 in V79-171 cells incubated in medium containing WR-2721. Int. J. Radiat. Biol. 1985, 47, 23–27. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Sivridis, E.; Maltezos, E.; Koukourakis, M.I. Down-regulation of intestinal-type alkaline phosphatase in the tumor vasculature and stroma provides a strong basis for explaining amifostine selectivity. Semin. Oncol. 2002, 29 (Suppl 19), 14–21. [Google Scholar] [CrossRef]
- Nagy, B.; Dale, P.J.; Grdina, D.J. Protection against cis-diamminedichloroplatinum cytotoxicity and mutagenicity in V79 cells by 2-[(aminopropyl)amino]ethanethiol. Cancer Res. 1986, 46, 1132–1135. [Google Scholar]
- Fenech, M.; Holland, N.; Chang, W.P.; Zeiger, E.; Bonassi, S. The Human Micronucleus Project – an international collaborative study on the use of the micronucleus technique for measuring DNA damage in humans. Mutat. Res. 1999, 428, 271–283. [Google Scholar] [CrossRef]
- Rudd, N.L.; Hoar, D.I.; Greentree, C.L.; Dimnik, L.S.; Hennig, U.G. Micronucleus assay in human fibroblasts: a measure of spontaneous chromosomal instability and mutagen hypersensitivity. Environ. Mol. Mutagen. 1998, 12, 3–13. [Google Scholar]
- Ramirez, M.H.; Ruppitsch, W.; Hirsch-Kauffmann, M.; Schweiger, M. Chromosomal instability of fanconi anemia cells is not the consequence of a defective repair activity of the ribosomal protein S3. Biochem. Biophys. Res. Commun. 1999, 264, 518–524. [Google Scholar] [CrossRef]
- Littlefield, L.G.; Joiner, E.E.; Colyer, S.P.; Sallam, F.; Frome, E.L. Concentration-dependent protection against X-ray-induced chromosome aberrations in human lymphocytes by the aminothiol WR-1065. Radiat. Res. 1993, 133, 88–93. [Google Scholar] [CrossRef]
- Blasiak, J.; Gloc, E.; Mlynarsky, W.; Drzewoski, J.; Skórski, T. Amifostine differentially modulates DNA damage evoked by idarubicin in normal and leukemic cells. Leuk. Res. 2002, 26, 1093–1096. [Google Scholar] [CrossRef]
- Buschini, A.; Alessandrini, C.; Martino, A.; Pasini, L.; Rizzoli, V.; Carlo-Stella, C.; Poli, P.; Rossi, C. Bleomycin genotoxicity and amifostine (WR-2721) cell protection in normal leukocytes vs. K562 tumoral cells. Biochem. Pharmacol. 2002, 63, 967–975. [Google Scholar] [CrossRef]
- Grdina, D.J.; Carnes, B.A.; Grahn, D.; Sigdestad, C.P. Protection against late effects of radiation by S-2-(3-aminopropylamino)-ethylphosphorothioic acid. Cancer Res. 1991, 51, 4125–4130. [Google Scholar]
- Kataoka, Y.; Basic, I.; Perrin, J.; Grdina, D.J. Antimutagenic effects of radioprotector WR-2721 against fission-spectrum neurons and 60Co gamma rays in mice. Int. J. Radiat. Biol. 1992, 61, 387–392. [Google Scholar] [CrossRef]
- Grdina, D.J.; Nagy, B.; Hill, C.K.; Wells, R.L.; Peraino, C. The radioprotector WR1065 reduces radiation-induced mutations at the hypoxanthine-guanine phosphoribosyl transferase locus in V79 cells. Carcinogenesis 1985, 6, 929–931. [Google Scholar] [CrossRef]
- Nagy, B.; Grdina, D.J. Protective effects of 2-[(aminopropyl)amino]ethanethiol against bleomycin and nitrogen mustard-induced mutagenicity in V79 cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1475–1478. [Google Scholar] [CrossRef]
- Shall, L.M.; Glover, D.; Turrisi, A.; Brown, D.Q.; Bonner, H.S.; Norfleet, A.L.; Weiler, C.; Glick, J.H.; Kligerman, M.M. Pharmacokinetics of WR-2721. Pharmacol. Therapeut. 1988, 39, 195–201. [Google Scholar] [CrossRef]
- Romanul, F.C.; Bannister, R.G. Localized areas of high alkaline phosphatase activity in endothelium of arteries. Nature 1962, 195, 611–612. [Google Scholar] [CrossRef]
- Mori, T.; Nikaido, O.; Suguhara, T. Dephosphorylation of WR-2721 with mouse tissue homogenates. Int. J. Radiat. Oncol. Biol. Phys. 1984, 10, 1529–1531. [Google Scholar] [CrossRef]
- Valeriote, F.; Tolen, S. Protection and potentiation of nitrogen mustard cytotoxicity by WR-2721. Cancer Res. 1982, 42, 4330–4331. [Google Scholar]
- Facorro, G.; Sarrasaque, M.M.; Torti, H.; Hager, A.; Avalos, J.S.; Foncuberta, M.; Kusminsky, G. Oxidative study of patients with total body irradiation: effects of amifostine treatment. Bone Marrow Transplant. 2004, 33, 793–798. [Google Scholar] [CrossRef]
- Pagano, G.; Manini, P.; Bagchi, D. Oxidative stress-related mechanisms are associated with xenobiotics exerting excess toxicity to Fanconi anemia cells. Environ. Health Perspect. 2003, 111, 1699–1703. [Google Scholar] [CrossRef]
- Mian, I.S.; Moser, M.J. The Fanconi anemia complementation group A protein contains a peroxidase domain. Mol. Genet. Metabol. 1998, 63, 230–234. [Google Scholar] [CrossRef]
- Kruyt, F.A.E.; Hoshino, T.; Liu, J.M.; Joseph, P.; Jaiswal, A.K.; Youssoufian, H. Abnormal microsomal detoxification implicated in Fanconi anemia group C by interaction of the FAC protein with NADPH cytochrome P450 reductase. Blood 1998, 92, 3050–3056. [Google Scholar]
- Cumming, R.C.; Lightfoot, J.; Beard, K.; Youssoufian, H.; O’Brien, P.J.; Buchwald, M. Fanconi anemia group C protein prevents apoptosis in hematopoietic cells through redox regulation of GSTP1. Nat. Med. 2001, 7, 814–820. [Google Scholar] [CrossRef]
- Futaki, M.; Igarashi, T.; Watanabe, S.; Kajigaya, S.; Tatsuguchi, A.; Wang, J.; Liu, J.M. The FANCG Fanconi anemia protein interacts with CYP2E1: possible role in protection against oxidative DNA damage. Carcinogenesis 2002, 23, 67–72. [Google Scholar] [CrossRef]
- Akkari, Y.M.; Bateman, R.L.; Reifsteck, C.A.; D’Andrea, A.D.; Olson, S.B.; Grompe, M. The 4N cell cycle delay in Fanconi anemia reflects growth arrest in late S phase. Mol. Genet. Metabol. 2001, 74, 403–412. [Google Scholar]
- North, S.; El-Ghissassi, F.; Pluquet, O.; Verhaegh, G.; Hainaut, P. The cytoprotective aminothiol WR-1065 activates p21waf-1 and down regulates cell cycle progression through a p53-dependent pathway. Oncogene 2000, 19, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Smoluk, G.D.; Fahey, R.C.; Ward, J.F. Equilibrium dialysis studies of the binding of radioprotector compounds to DNA. Radiat. Res. 1986, 107, 194–204. [Google Scholar] [CrossRef]
- Murley, J.S.; Constantinou, A.; Kamath, N.S.; Grdina, D.J. WR-1065, an active metabolite of the cytoprotector amifostine, affects phosphorylation of topoisomerase II alpha leading to changes in enzyme activity and cell cycle progression in CHO cells. Cell Proliferat. 1997, 30, 283–294. [Google Scholar] [CrossRef]
- Shen, H.; Chen, Z.J.; Zilfou, J.T.; Hopper, E.; Murphy, M.; Tew, K.D. Binding of the aminothiol WR-1065 to transcription factors influences cellular response to anticancer drugs. Pharmacol. Exp. Ther. 2001, 297, 1067–1073. [Google Scholar]
- Cohen-Haguenauer, O.; Peault, B.; Bauche, C.; Daniel, M.T.; Casal, I.; Levy, V.; Dausset, J.; Boiron, M.; Auclair, C.; Gluckman, E.; Marty, M. In vivo repopulation ability of genetically corrected bone marrow cells from Fanconi anemia patients. PNAS 2006, 103, 2340–2345. [Google Scholar] [CrossRef]
- Jänne, J.; Alhonen, L.; Pietila, M.; Keinanen, T.A. Genetic approaches to the cellular functions of polyamines in mammals. Eur. J. Biochem. 2004, 271, 877–894. [Google Scholar]
- Korkina, L.G.; Samochatova, E.V.; Maschan, A.A.; Suslova, T.B.; Cheremisina, Z.P.; Afanasev, I.B. Release of active oxygen radicals by leukocytes of Fanconi anemia patients. J. Leukocyte Biol. 1992, 52, 357–362. [Google Scholar]
- Pagano, G.; Korkina, L.G. Prospects for nutritional interventions in the clinical management of Fanconi anemia. Cancer Cause Control 2000, 11, 881–889. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Camelo, R.M.; Kehdy, F.S.G.; Salas, C.E.; Lopes, M.T.P. Amifostine Protection Against Mitomycin-induced Chromosomal Breakage in Fanconi Anaemia Lymphocytes. Molecules 2008, 13, 1759-1772. https://doi.org/10.3390/molecules13081759
Camelo RM, Kehdy FSG, Salas CE, Lopes MTP. Amifostine Protection Against Mitomycin-induced Chromosomal Breakage in Fanconi Anaemia Lymphocytes. Molecules. 2008; 13(8):1759-1772. https://doi.org/10.3390/molecules13081759
Chicago/Turabian StyleCamelo, Ricardo M., Fernanda S. G. Kehdy, Carlos E. Salas, and Miriam T. P. Lopes. 2008. "Amifostine Protection Against Mitomycin-induced Chromosomal Breakage in Fanconi Anaemia Lymphocytes" Molecules 13, no. 8: 1759-1772. https://doi.org/10.3390/molecules13081759
APA StyleCamelo, R. M., Kehdy, F. S. G., Salas, C. E., & Lopes, M. T. P. (2008). Amifostine Protection Against Mitomycin-induced Chromosomal Breakage in Fanconi Anaemia Lymphocytes. Molecules, 13(8), 1759-1772. https://doi.org/10.3390/molecules13081759