Rigorous Biogenetic Network for a Group of Indole Alkaloids Derived from Strictosidine

Abstract
:Introduction
- Compounds isolated from the same species reveal their common origin. Which alkaloids could be isolated from those plant species which produce also strictosidine (3)? In other terms: which are the “coalkaloids” of this universal precursor of indole alkaloids?
- The monomer components necessarily coexist in the same species, from which the dimer alkaloids were isolated. This fact likewise reveals chemotaxonomic connections among the individual alkaloids and species. Which types of monomer components are connected in the dimers?

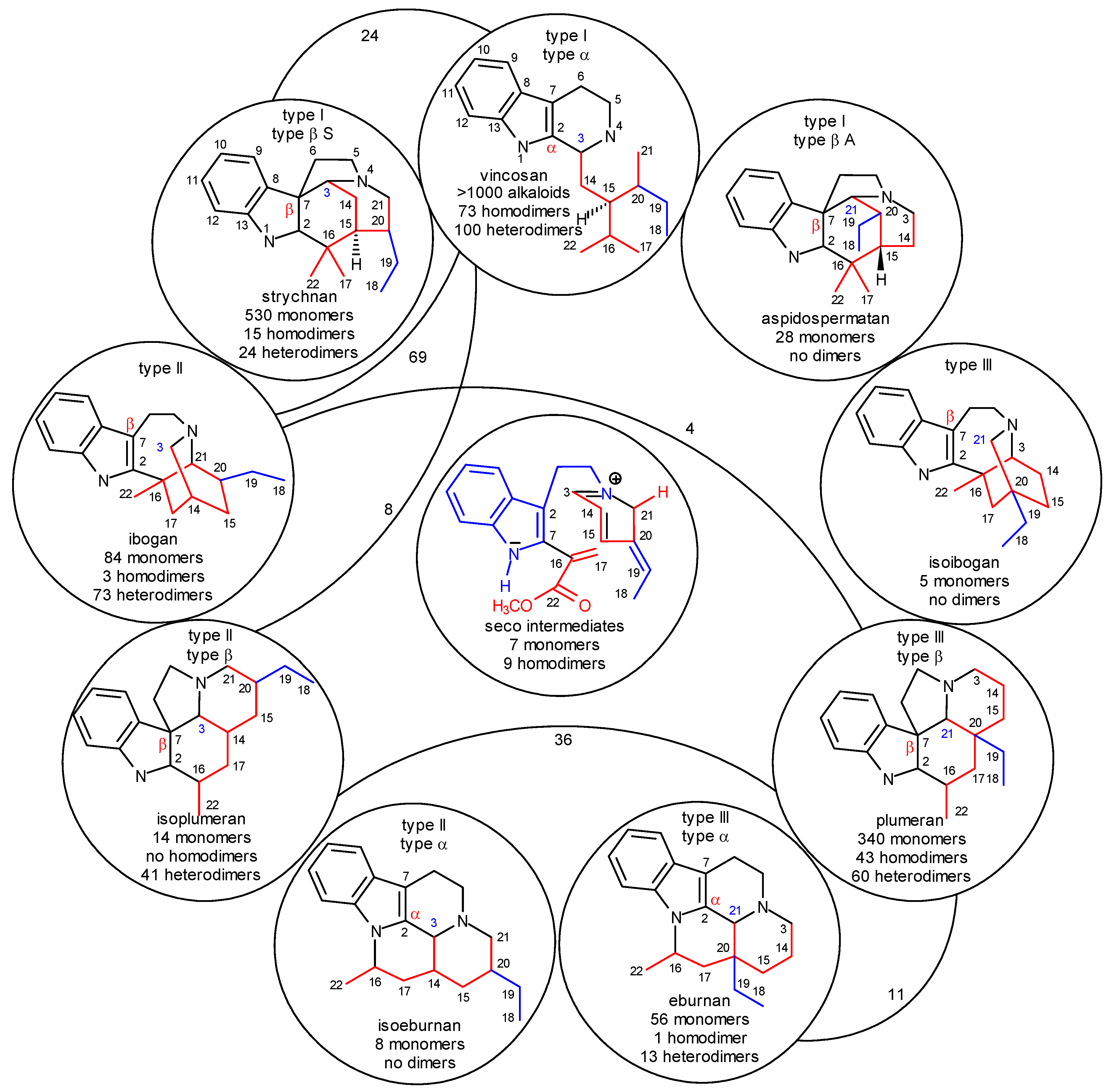
The Biogenetic-type System of Indole Alkaloids

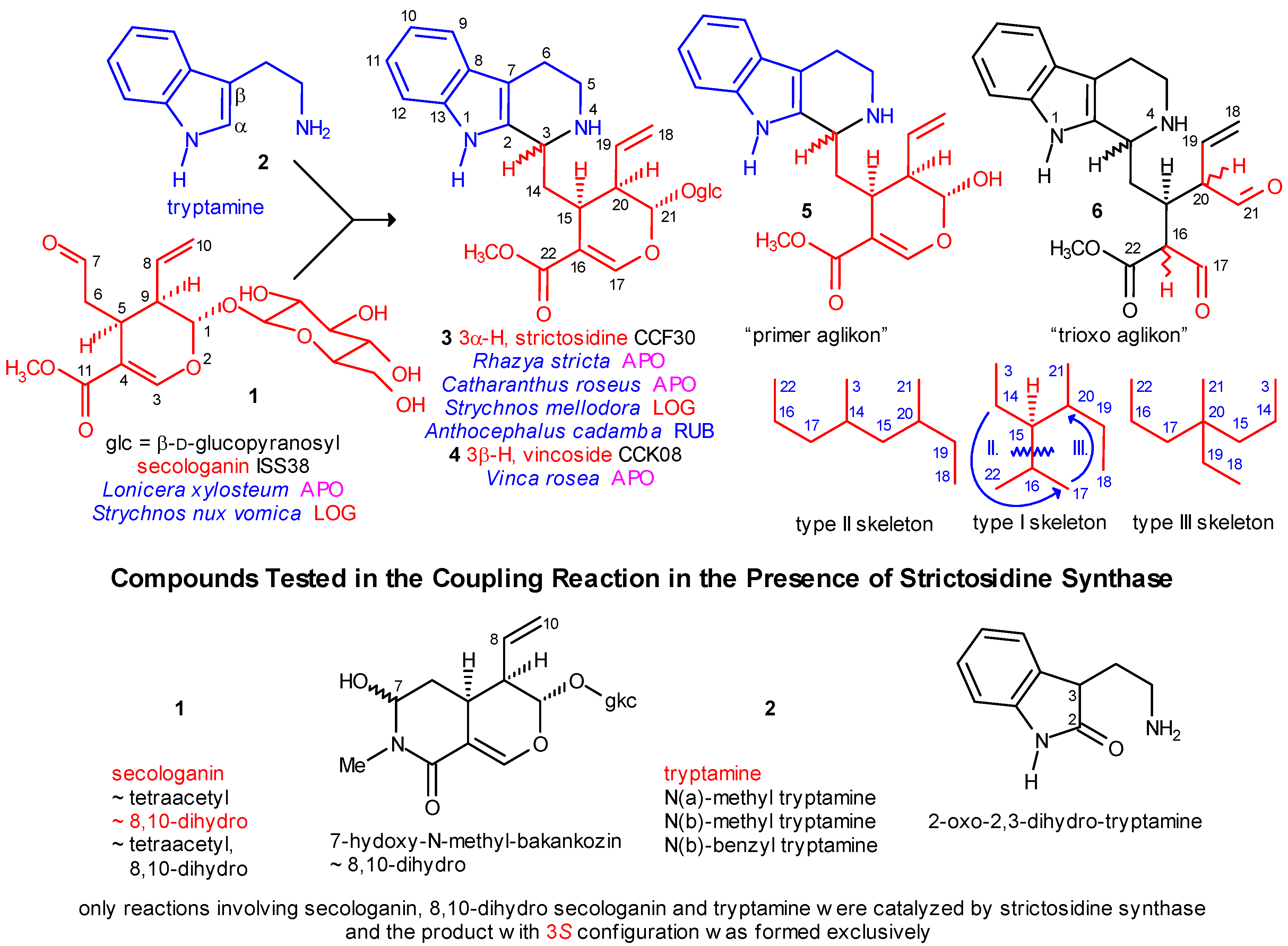
- level 0:
- formation of strictosidine (3) from secologanin and tryptamine and removal of the glucosyl subunit (Scheme 1);
- level 1:
- cyclization of the secologanin subunit to N-1 or/and N-4 of the tryptamine subunit (Scheme 2 shows only cyclizations to N-4);
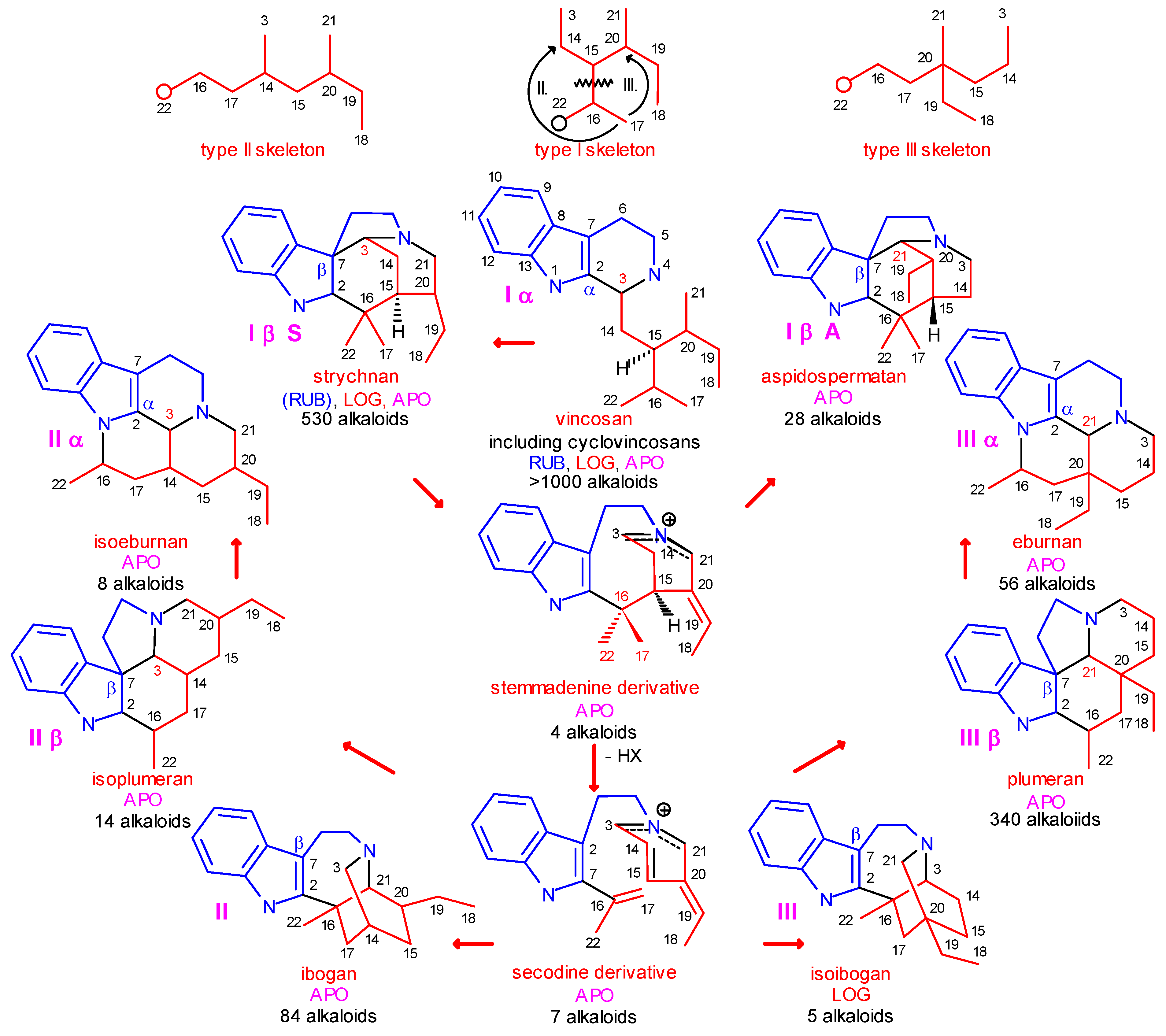
- level 2:
- in the secologanin subunit, transformation of the type I skeleton into the type II and type III ones (Scheme 1);
- level 3:
- attachment of C-3 or C-21 or neither of them to C-2 (α) or C-7 (β) position of the the indole ring;
- level 4:
- further cyclizations between the secologanin and the tryptamine subunits;
- level 5:
- further cyclizations inside the secologanin subunit;
- level 6:
- further transformations (e.g. rearrangements, ring extensions and ring contractions; fragmentation of the tryptamine side chain, formation of sesqui- and dimers, which are temporarily omitted).
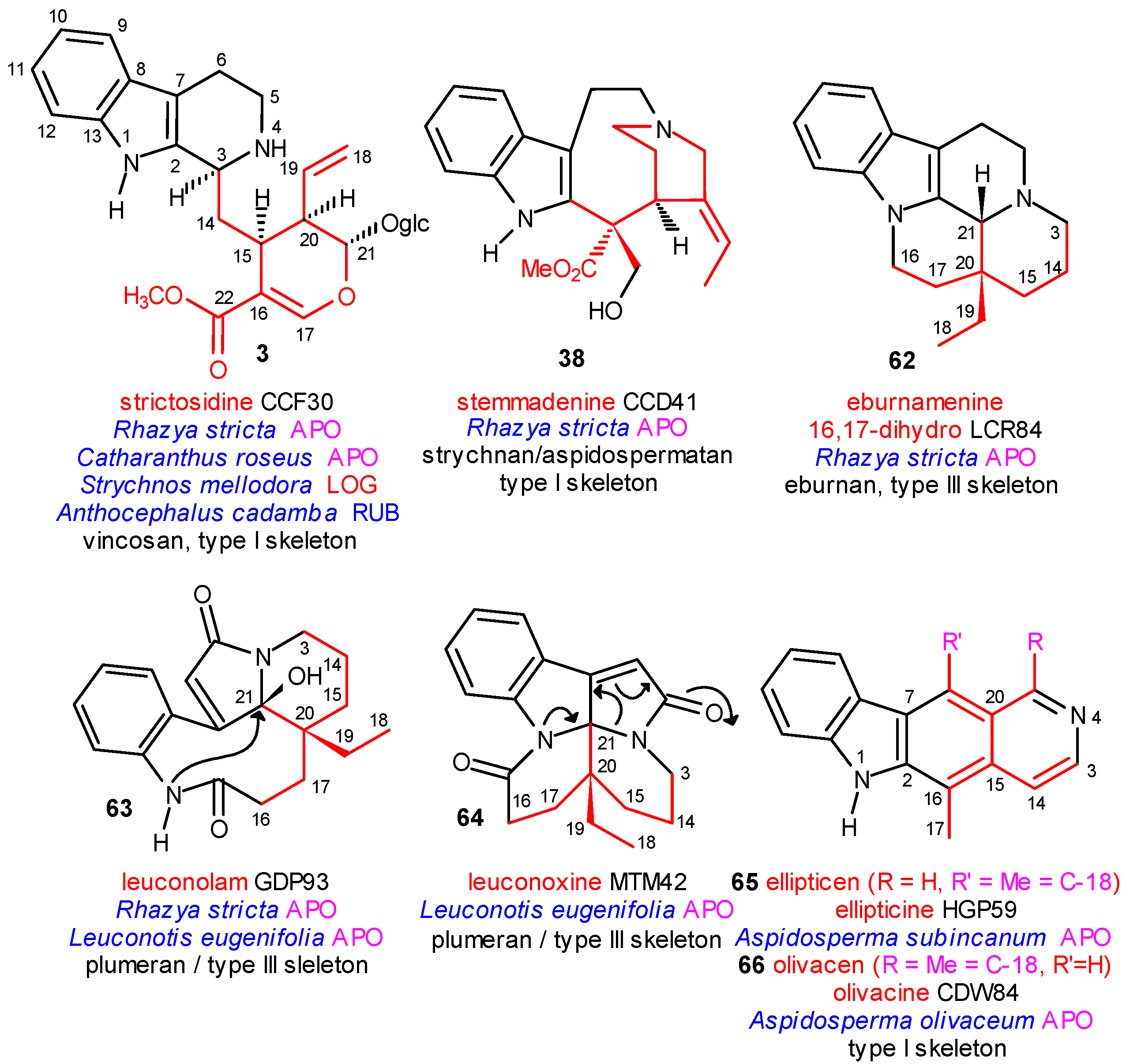
The Main Precursor Strictosidine

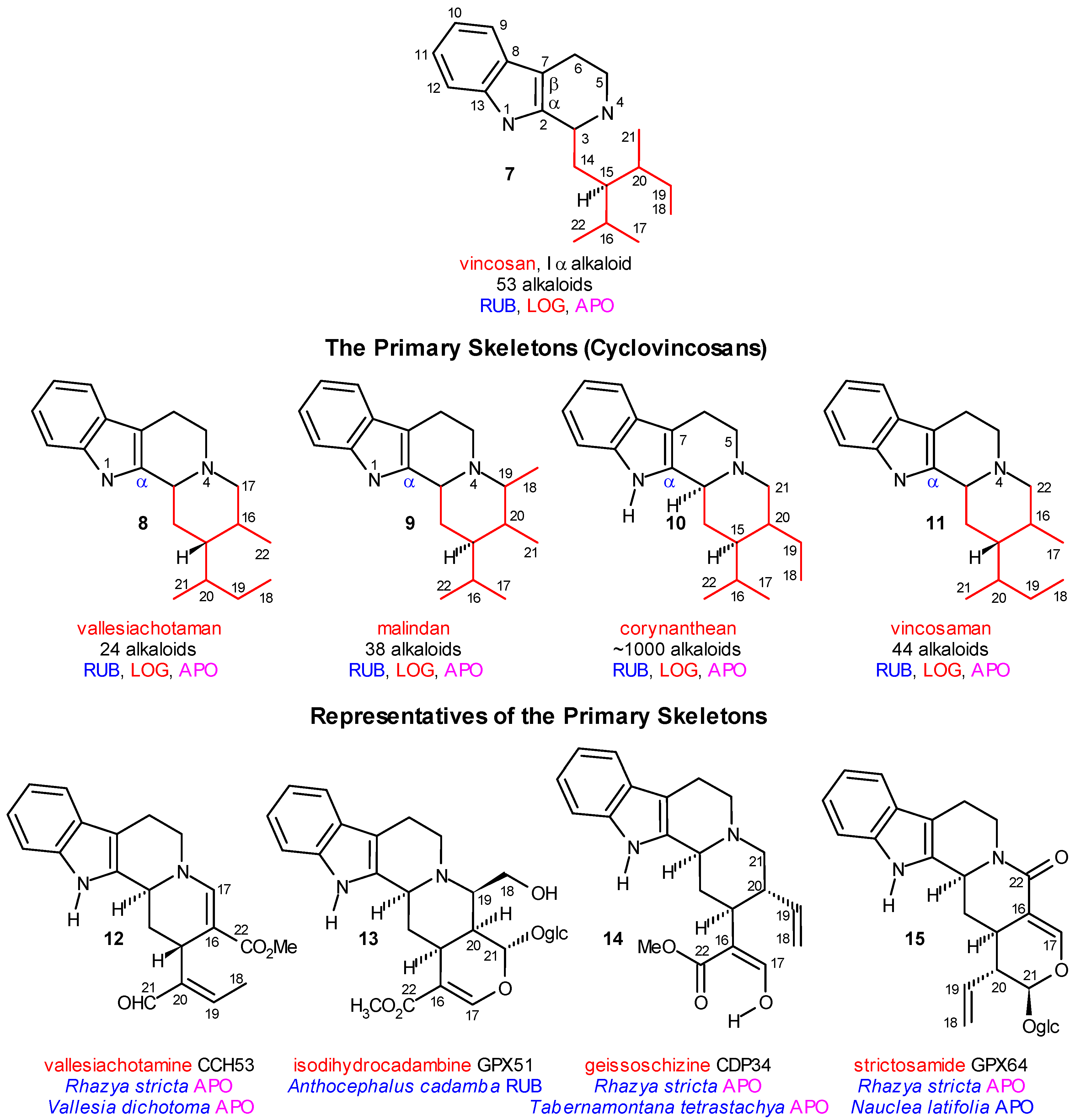
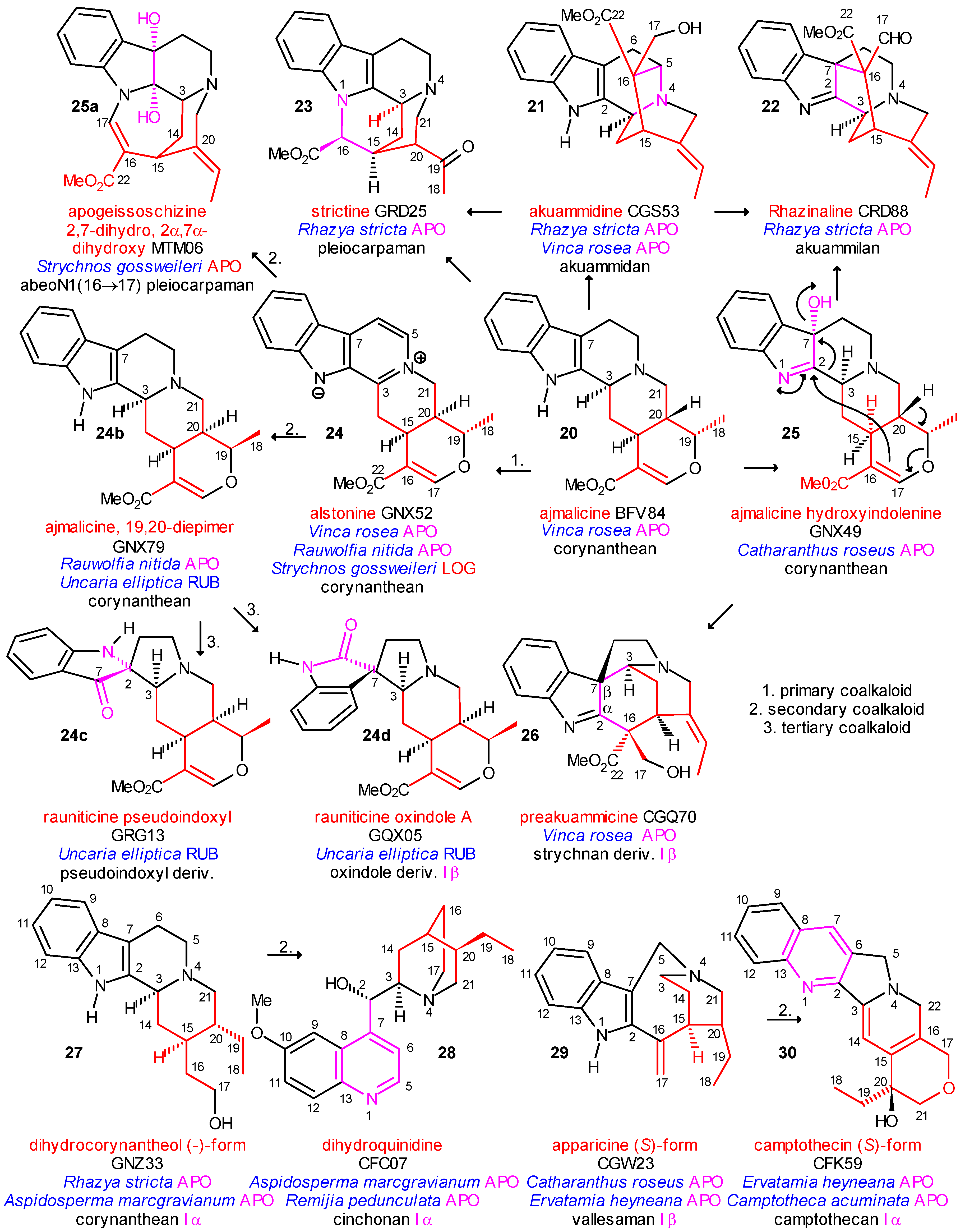
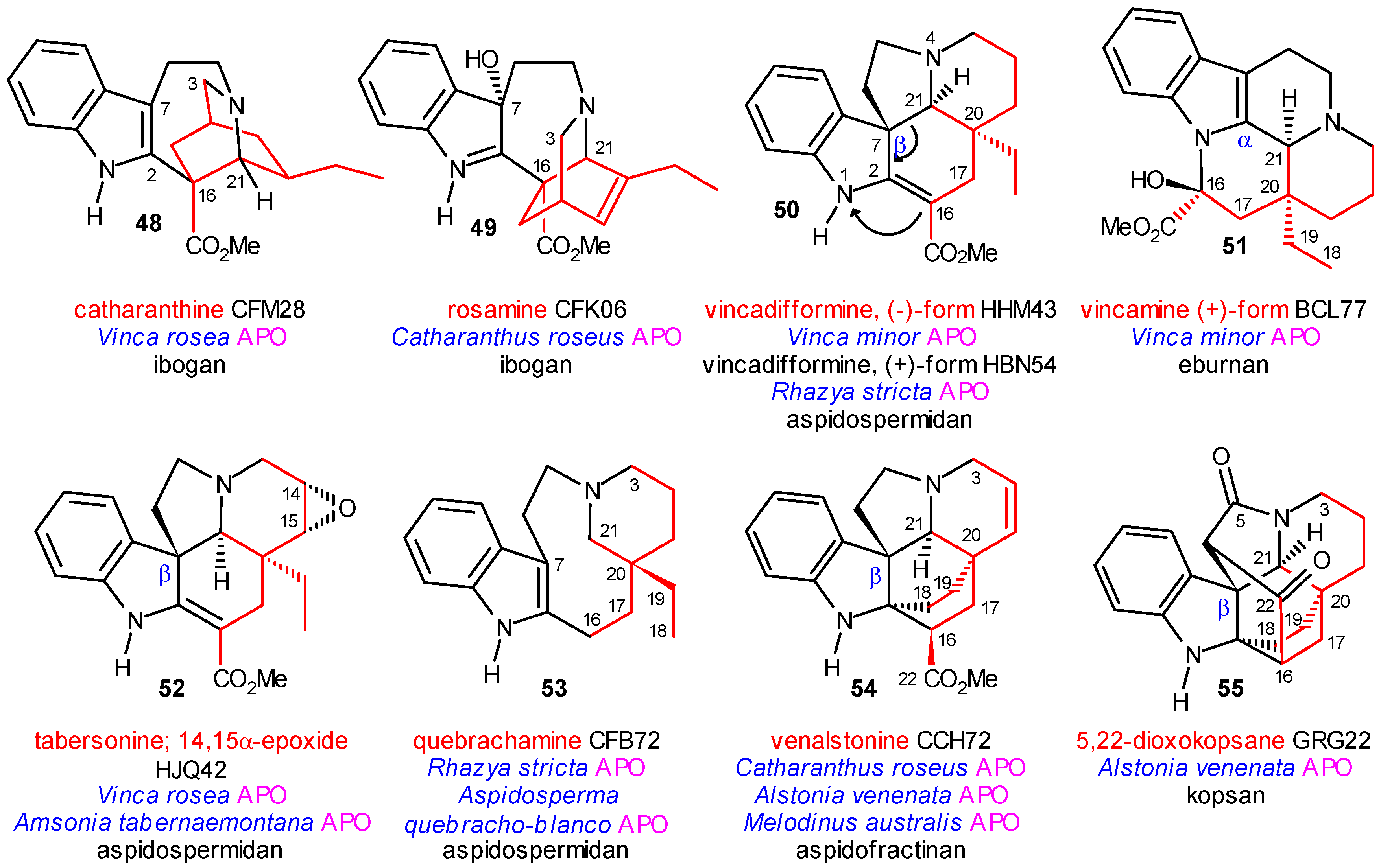
Coalkaloids of Strictosidine in Type I α Class


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
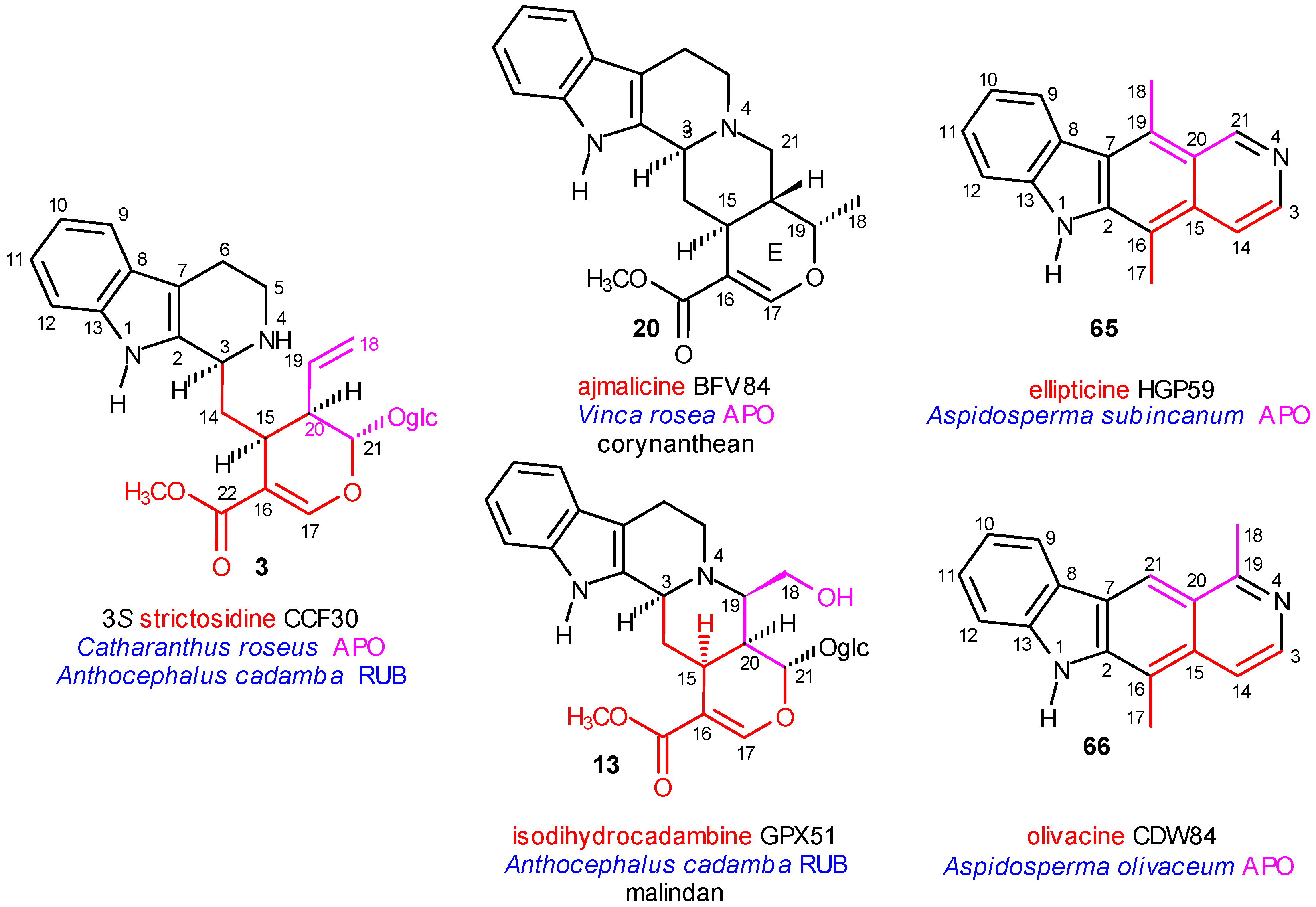{kind=link}
| Anthocephalus cadamba (RUB) (8 alkaloids) | Rhazya stricta (APO) (50 alkaloids) |
| Cadambine, CFJ 83, isomalindan | Akuammicine deriv. GNP98, strychnan |
| Cadamine, 2 derivs. HFP34, GZM28, malindan | Akuammidine CGS53, akuammidan |
| Dihydrocadambine, 2 derivs. GPX71, GPX73, isomalindan | Akuammiline deriv. CGS78, akuammilan |
| Isodihydrocadambine, 2 derivs. GPX51, GPX53, malindan | Anthirine deriv. CGV97, anthiran |
| Strictosidine, CCF30, vincosan | Aspidospermidine 2 derivs. NSG56, NGX40, plumeran |
| Bharhingine GRD15, strychnan | |
| Catharanthus roseus (= Vinca Rosea) (APO) (49 alkaloids) | Burnamine deriv. CGP15, akuammilan |
| Ajmalicine deriv. BFV84 (VR), GNX93, corynanthean | Decarboxymethoxytetrahydrosecodine deriv NQD29 secodan |
| Ajmalicine hydroxyindolenine deriv. GNX49, corynanthean | Dihydrocorynantheol deriv. GNZ33, corynanthean |
| Akuammicine deriv. CGS34, strychnan | Dihydroeburnamenine LCR84, eburnan |
| Akuammiline deriv. LTT85, akuammilan | Eburenine 2 derivs. GZP25, LNJ42, plumeran |
| Alioline, OSQ04, ibogan | Geissoschizine CDP34, corynanthean |
| Alstonine 2 derivs. GNX52 (VR), CCC44, corynanthean | Isorhazicine BQR50, secoajmalan |
| Apparicine (S)-form, CGW23, vallesaman | Isositsirikine 4 derivs. LCS06, CCW87, NBV19, NBV18, corynanthean |
| Bannucine CQG04, plumeran | Lanceomigine deriv. CHL97, akuammilan |
| Catharanthine CFM58 ibogan (VR) | Leuconolam NNZ31, plumeran |
| Fluorocarpamine indoxyl deriv. GRG17 pleiocarpaman | Nor-C-fluorocurarine deriv. HHZ45, strychnan |
| Isositsirikine 3 derivs. CCW78 (VR), CCW86, CCW87, corynanthean | Quebrachamine deriv. CFB72, plumeran |
| Lochneridine BQS00 (VR) strychnan | Rhazidigenine hydroxyindolenine 2 derivs. CFF52, CFF54, plumeran |
| Perivine 2 derivs. CGN81 (VR), CGN86 (VR), vobasan | Rhazimine 2 (quinoline) derivs. CFK02, CFK05, FYL18, akuammilan |
| Preakuammicine CGQ70 (VR), strychnan | Rhazinaline 2 derivs. CDR88, NXF68, akuammilan |
| Rosicine CFK07, plumeran/isoplumeran | Rhazinilam 2 derivs. GPF00, FNO12, plumeran |
| Sarpagin deriv BCC19 (VR) akuammidan | Rhazizine LDC76, strychnan |
| Sitsirikine 2 derivs. HJQ31 (VR), BCF50 (VR), corynanthean | Secodine 2 derivs. CHM24, BFV95, secodan |
| Strictamine deriv. CFF61, akuammilan | Stemmadenine CCD41, strychnan/aspidospermatan |
| Strictosidine CCF30 (+VR), vincosan | Strictamine 3 derivs. HHT53, CDR79, BFY65, akuammilan |
| Strictosidine 2 derivs. CCK08 (VR), CCK10 (VR) | Strictanine BQS42, plumeran |
| Tabersonine 2 derivs. GQZ42, HJQ42 (VR), plumeran | Stricticine GRD24, GRG17, plumeran |
| Talpinine deriv. GQW95, akuammidan | Strictine GRD25, pleiocarpaman |
| Tombozine deriv. GRC35 (+VR), akuammidan | Strictosamide GPX64, vincosaman |
| Venalstonine deriv. CCH72, plumeran | Strictosidine CCF30, vincosan |
| Vincadifformine deriv. CCJ46 (VR), plumeran | Vallesiachotamine 3 derivs. CCH54, CCH55, LXQ87, anthiran |
| Vincarodine GNK80, eburnan | Vincadifformine 3 derivs. HBN54, LHX61, CCJ49, plumeran |
| Vincoline CCK01 (VR), plumeran | Vincamajine deriv. LJB72, akuammidan |
| Vindolidine 5 derivs. KLJ88 (+VR), JHQ68, JRJ50 GQZ38 GQZ39, plumeran | |
| Vindolinine 4 derivs. BQX53 (+VR), MVY20, BQX56, BQX57, plumeran | Strychnos mellodora (LOG) (5 alkaloids) |
| Voalutein indoxyl deriv. CFK06, ibogan | Lyaloside 2 derivs. GVY09, GVY10, vincosan |
| (VR) indicates alkaloids isolated (also) from Vinca rosea | Strictosidine CCF30, vincosan |
| Strictosidine 2 derivs. LHX56, MTN39, vincosan |

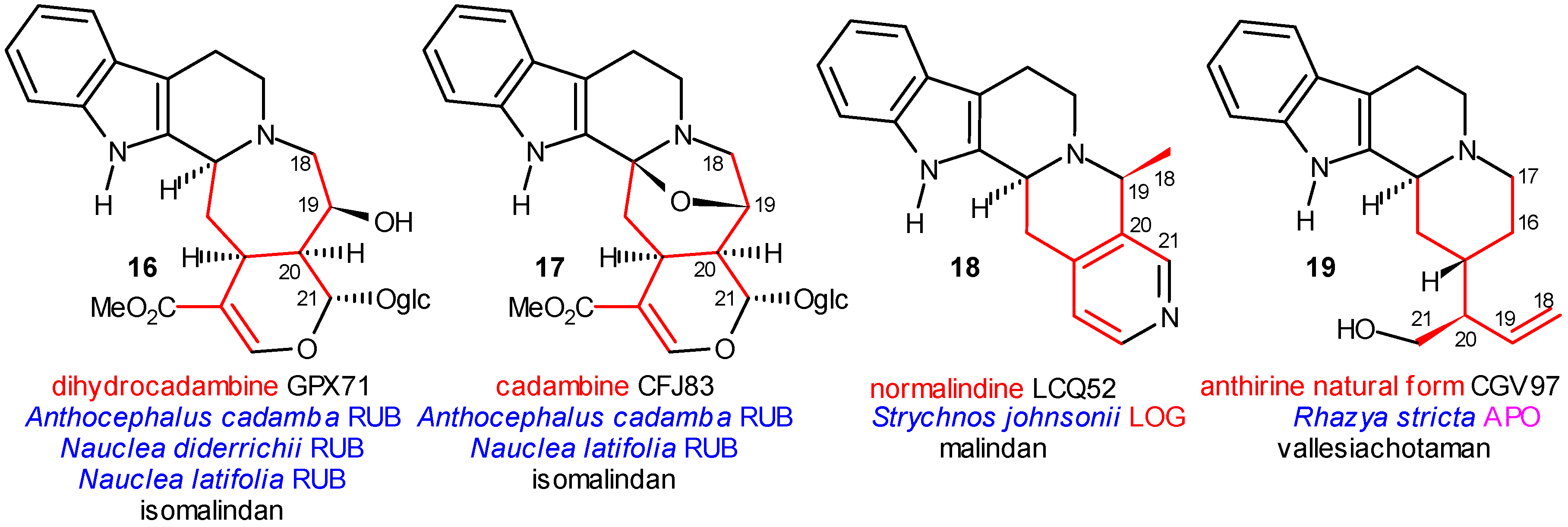
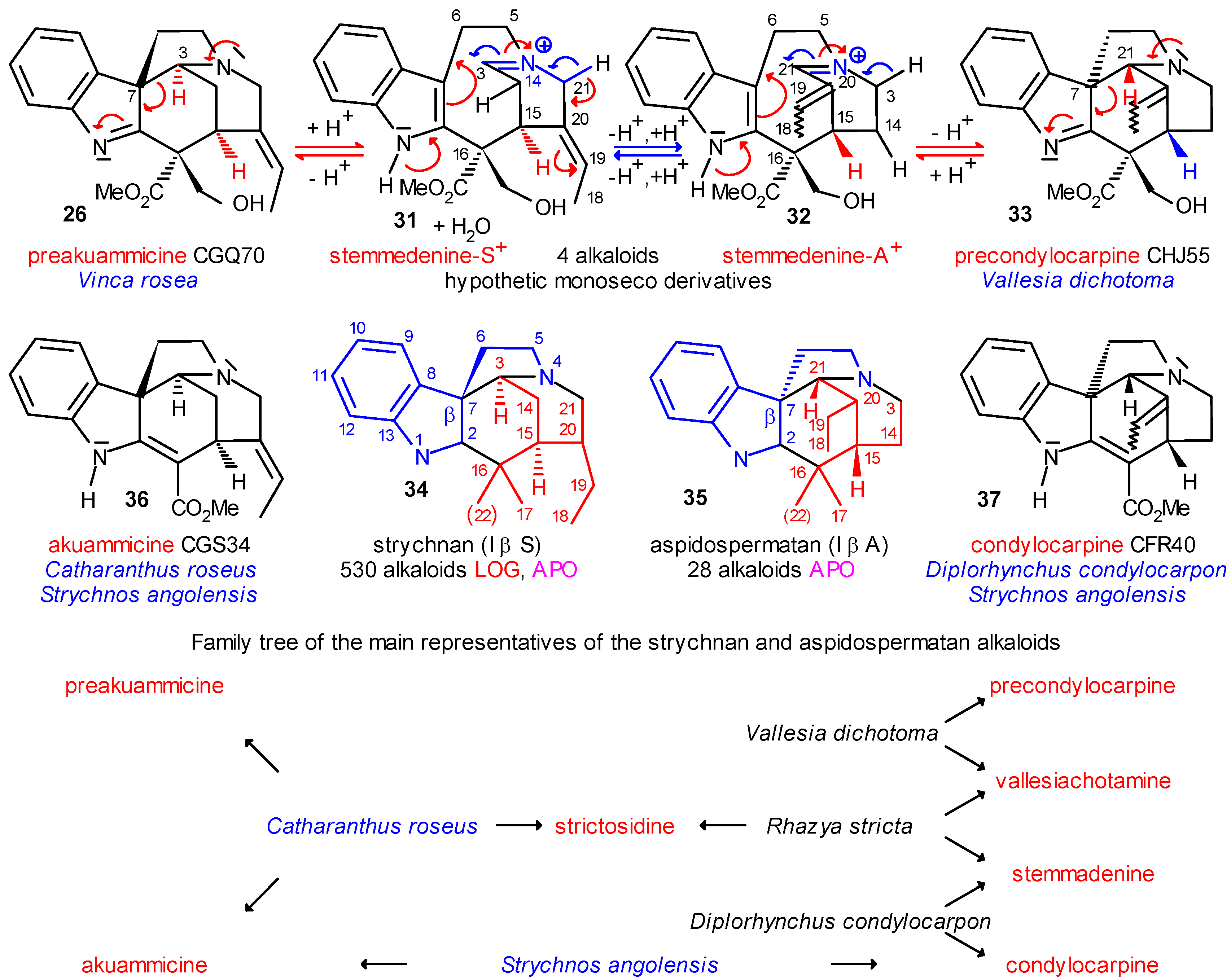
Coalkaloids of Strictosidine in Type I β Class

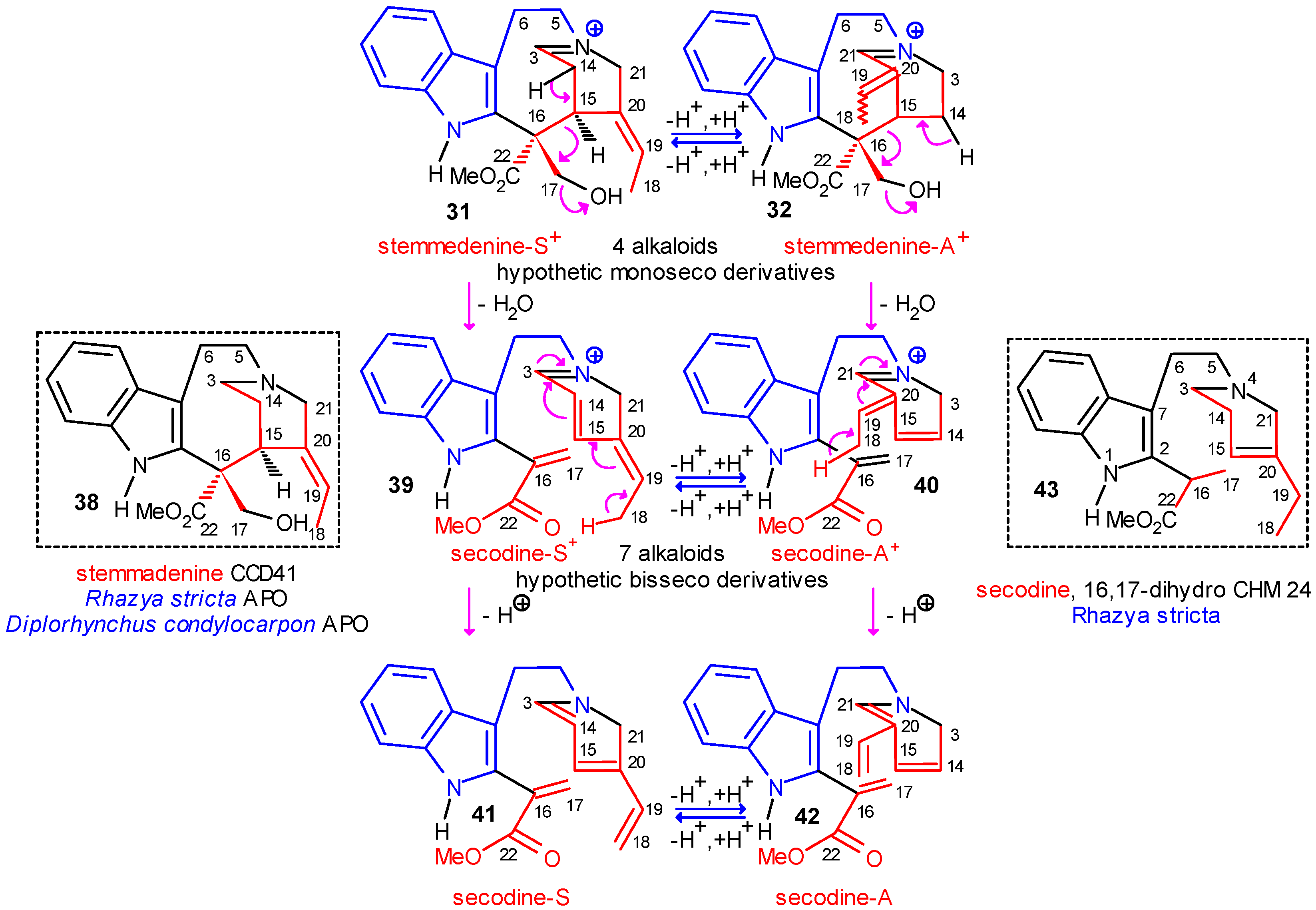
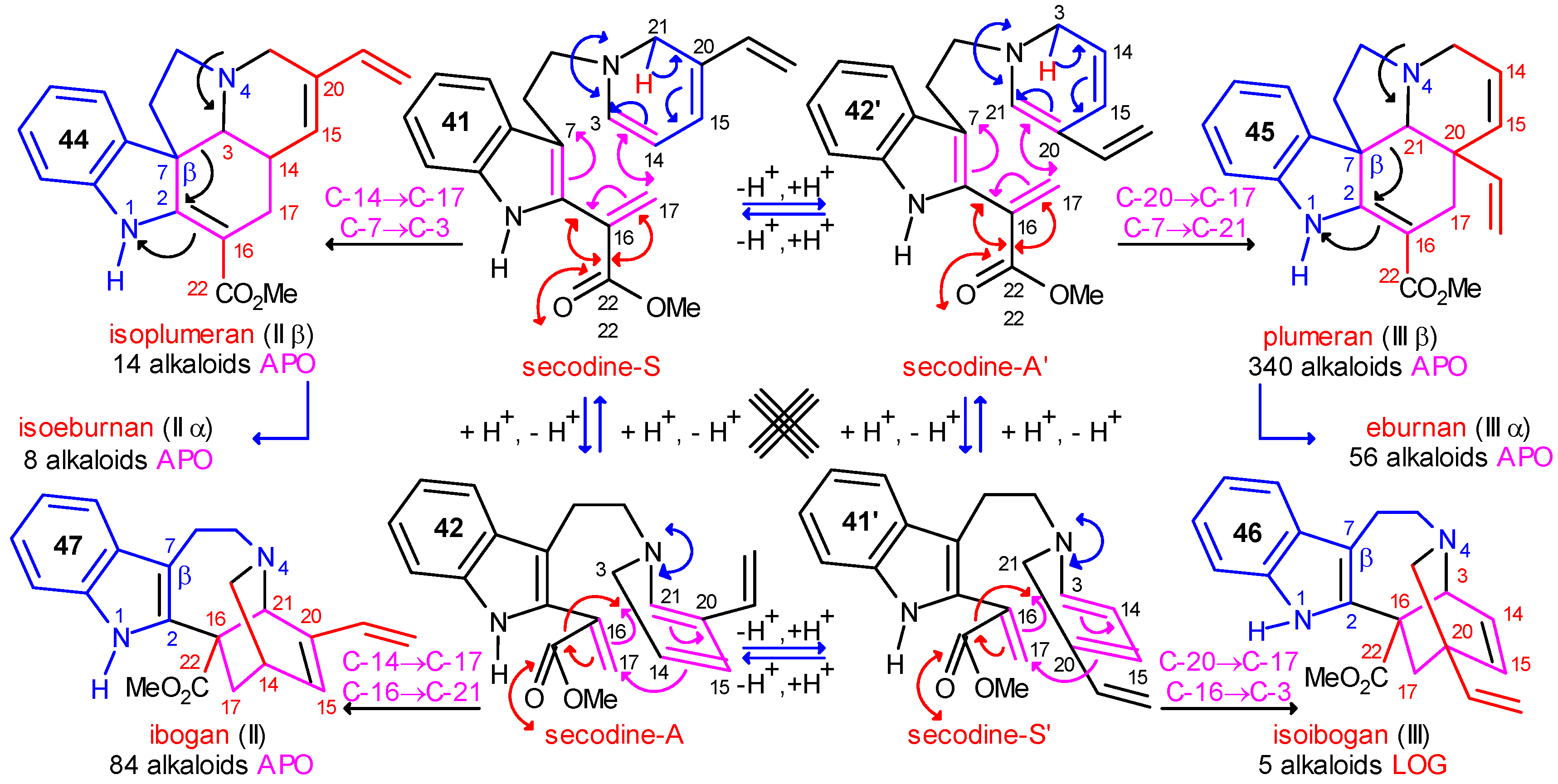
The Central Role of Stemmadenine and Secodine Derivatives


 position of the indole ring (arrows in the formulas) and give in several steps the eburnan (III α) and isoeburnan (II α) alkaloids (see in Scheme 2), respectively. This would be the last phase in the molecular evolution of the indole alkaloids. Finally, it should be emphasized that the transition from the type I to the type II/III class of indole alkaloids is possible only in the evolution line of the corynanthean skeleton.
position of the indole ring (arrows in the formulas) and give in several steps the eburnan (III α) and isoeburnan (II α) alkaloids (see in Scheme 2), respectively. This would be the last phase in the molecular evolution of the indole alkaloids. Finally, it should be emphasized that the transition from the type I to the type II/III class of indole alkaloids is possible only in the evolution line of the corynanthean skeleton.Coalkaloids of Strictosidine in Type II and Type III Classes


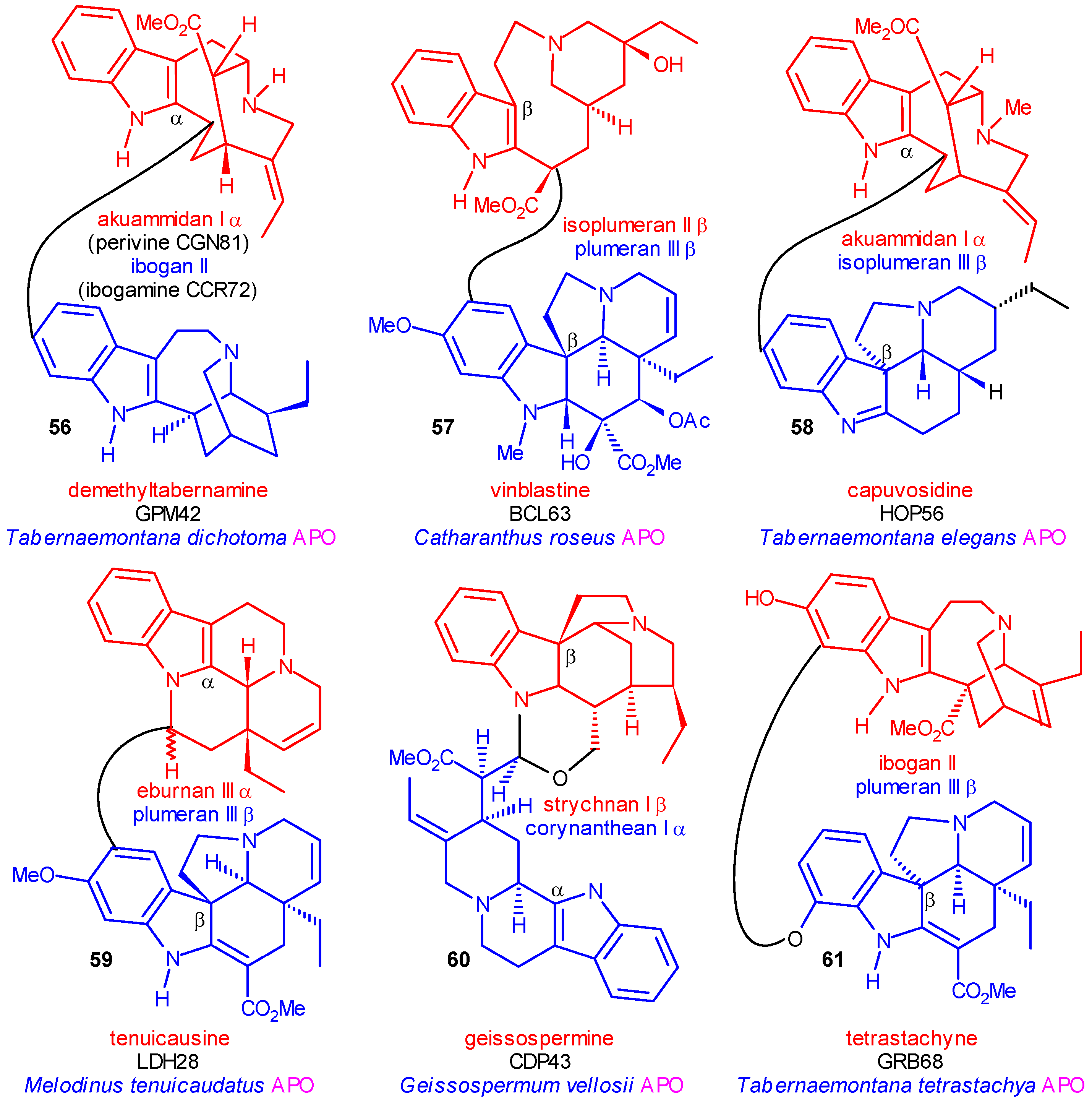
The Significance of Dimer Alkaloids in the Biogenesis of Indole Alkaloids

Outlook


Summary
 |
Acknowledgements
References
- Cordell, G. A. The Biosythesis of Indole Alkaloids. Lloydia 1974, 37, 219–298. [Google Scholar]
- Tietze, L.-F. Secologanine, eine biogenetische Schlüsselverbindung – Synthese und Biogenese der Iridoid- und Secoiridoidglykoside. Angew. Chem. 1983, 93, 840–853. [Google Scholar]
- Atta-Ur-Rahman; Anwer, B. Biosynthesis of Indole Alkaloids; Clarendon Press: Oxford, United Kingdom, 1983. [Google Scholar]
- Stoeckigt, J.; Ruppert, L. Strictosidine, the Biosynthetic Key to Monoterpenoid Indole Alkaloids. In Comprehensive Natural Products Chemisty; Kelly, J.W., Ed.; Elsevier B. V.: Amsterdam, The Netherlands, 1999; Vol. 4, pp. 109–138. [Google Scholar]
- Talayama, H.; Sakai, S.-I. Monoterpenoid Indole Alkaloids Syntheses Utilizing Biomimetic Reactions. In The Alkaloids; Cordell, J.W., Ed.; Academic Press: New York, NY, USA, 1998; Volume 50, pp. 415–452. [Google Scholar]
- Verpoorte, R.; van der Heijden, R.; Moreno, P.R. Biosynthesis of Terpenoid Indole Alkaloids in Catharanthus roseus Cells. In The Alkaloids; Cordell, J.W., Ed.; Academic Press: New York, NY, USA, 1997; Volume 49, pp. 221–298. [Google Scholar]
- El-hayed, K.; Verpoorte, R. Catharanthus Terpenoid Alkaloids: Biosynthesis and Regulations. Phytochem. Rev. 2007, 6, 277–305. [Google Scholar] [CrossRef]
- Stoeckigt, J.; Barleben, K.; Panjukar, S.; Loris, E.A. A 3-D Structure and Function of Strictosidine Synthase – the Key Enzyme of Monoterpenoid Indole Alkaloid Biosynthesis. Plant Physiol. Biochem. 2008, 46, 340–355, Erratum: ibid. 615. [Google Scholar] [CrossRef]
- Szabó, L.F. Some Aspects of the Chemisty of Indole Alkaloids. In Studies in Natural Products Chemistry; Vol. 26. Bioactive Natural Products. Part G.; Atta-Ur-Rahman, Ed.; Elsevier B. V.: Amsterdam, The Netherlands, 2002; pp. 95–148, a) Ibid. p. 132; b) Ibid. pp. 137-141. [Google Scholar]
- Patthy-Lukáts, Á.; Károlyházy, L.; Szabó, L.F.; Podányi, B. The First Direct and Detailed Stereochemical Analysis of Strictosidine. J. Nat. Prod. 1997, 60, 69–75. [Google Scholar] [CrossRef]
- Beke, Gy.; Károlyházy, L.; Patthy-Lukáts, Á.; Podányi, B.; Szabó, L. F. Chirality Transfer in the Formation of the Indole Alkaloids Derived from Secologanin. In Progress in Biological Chirality; Pályi, G., Zucchi, C., Cagloti, L., Eds.; Elsevier B. V.: Amsterdam, The Netherlands, 2004; pp. 377–395, (Chemotaxonomy in Indole Alkaloids. Part 1); a) Ibid. pp. 386-387; b) Ibid. pp. 389-392. [Google Scholar]
- Szabó, L.F. Diversity and Selectivity in Molecular Evolution. In Fundamentals of Life; Pályi, G., Zucchi, C., Caglioti, L., Eds.; Elsevier Life Sci.: Paris, France, 2002; pp. 437–449. [Google Scholar]
- Szabó, L.F. Reaction Mechanism and Chemotaxonomy in the Formation of the Type I Indole Alkaloids Derived from Secologanin. J. Phys. Org. Chem. 19, 2006, 579–591, (Chemotaxonomy in Indole alkaloids. Part 2); a) Ibid. pp. 582-586; b) Ibid. pp. 589-591. [Google Scholar]
- Szabó, L.F. Molecular Interrelations in the Melodinus Alkaloids. ARKIVOC 2007, 280–290, (Chemotaxonomy in Indole Alkaloids. Part 3). [Google Scholar]
- Szabó, L.F. Molecular Evolutionary Lines in the Formation of Indole Alkaloids Derived from Secologanin. ARKIVOC 2008, 167–181, (Chemotaxonomy in Indole Alkaloids. Part 4). [Google Scholar] [CrossRef]
- Buckingham, J. (Ed.) Dictionary of Natural Products on DVD, Version 16.2, Chapman and Hall/CRC: Boca Raton, FL, USA, 2008.
- Le Men, J.; Taylor, W.I. Uniform Numbering System for Indole Alkaloids. Experientia 1965, 21, 508–510. [Google Scholar]
- Kisakürek, M.V.; Leeuwenberg, A.J.; Hesse, M. A Chemotaxonomic Investigation of the Plant Families of Apocynaceae, Loganiaceae, and Rubiaceae by their Alkaloid Content. In Alkaloids: Chemistry and Biological Properties; Vol. 1, Pelletier, S.W., Ed.; John Wiley and Sons: New York, NY, 1983; pp. 211–376. [Google Scholar]
- Hesse, M. Alkaloids; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2002; pp. 237–256. [Google Scholar]
- Károlyházy, L.; Patthy-Lukáts, Á.; Szabó, L.F.; Podányi, B. Preparation of Recombinant Strictosidine Synthase Enzyme and Investigation of its Substrate Specifity. Gyógyszerészet 1995, 39, 447–448, [Chem. Abstr. 1995, 123, 221527]. [Google Scholar]
- Treimer, J.F.; Zenk, M.H. Purification and Properties of Strictosidine Synthase, the Key Enzyme in Indole Alkaloid Formation. Eur. J. Biochem. 1979, 101, 225–233. [Google Scholar] [CrossRef]
- Bracher, D.; Kutchan, T.M. Strictosidine Synthase from Rauwolfia serpentina: Analysis of a Gene Involved in Indole Alkaloid Biosynthesis. Arch. Biochem. Biophys. 1992, 294, 717–723. [Google Scholar] [CrossRef]
- Károlyházy, L.; Patthy-Lukáts, Á.; Szabó, L.F. Chemistry of Secologanin. Part 5. Graphical Analysis of the Acidic Deglycosylation of Vincoside Derivatives. J. Phys. Org. Chem. 1998, 11, 622–631. [Google Scholar] [CrossRef]
- Saunders, G.N.; Purdy, J.R.; McLean, S. Selective Transformation of Secologanin: Epoxidation. Can. J. Chem. 1983, 61, 276–281. [Google Scholar] [CrossRef]
- Shellard, E.; Houghton, P.J. Conversion of Pseudo Heteroyohimbine Alkaloids to Oxindoles. I. In vitro studies. Planta Med. 1971, 20, 167–171. [Google Scholar] [CrossRef]
- Finch, N.; Taylor, W.I. Oxidative Transformations of Indole Alkaloids. 1. Preparation of Oxindoles from Yohimbine Structure and Partial Syntheses of Mitraphylline, Rhincophylline, and Corynoxeine. J. Am. Chem. Soc. 1962, 84, 3871–3877. [Google Scholar] [CrossRef]
- Finch, N.; Gemenden, C.W.; Hsu, I.H.; Kerr, A.; Sim, G.A.; Taylor, W.I. Oxidative Transformations of Indole Alkaloids. 3. Pseudoindoxyls from Yohimbinoid Alkaloids and their Conversion to “Invert” Alkaloids. J. Am. Chem. Soc. 1965, 87, 2229–2235. [Google Scholar] [CrossRef]
- Károlyházy, L.; Patthy-Lukáts, Á.; Szabó, L.F.; Podányi, B. Chemistry of Secologanin. Part 7. Chemical Fragmentation of Secologanin: Biomimetic Transition from the Aliphatic into the Aromatic Skeleton. Tetrahedron Lett. 2000, 41, 1575–1578. [Google Scholar] [CrossRef]
- Tóth, F.; Oláh, J.; Kalaus, G.; Greiner, I.; Szőllősy, Á.; Gömöri, Á.; Hazai., Á.; Szántay, C. Synthesis of Vinca Alkaloids and Related Compounds. Part 110. A New Synthetic Method for Preparation of Pandoline-type Alkaloid-like Molecule. Tetrahedron. (accepted for publication).
- Hugel, G.; Massiot, G.; Levy, J.; Le Men, J. Methylenindolines, indolenines, and indoleniniums. XIII. 16-Hydroxy-1-dehydrovincadifformine, the Intermediate in the Biomimetic Rearrangement of Vincadifformine to Vincamine. Tetrahedron 1981, 37, 1369–1375. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szabó, L.F. Rigorous Biogenetic Network for a Group of Indole Alkaloids Derived from Strictosidine. Molecules 2008, 13, 1875-1896. https://doi.org/10.3390/molecules13081875
Szabó LF. Rigorous Biogenetic Network for a Group of Indole Alkaloids Derived from Strictosidine. Molecules. 2008; 13(8):1875-1896. https://doi.org/10.3390/molecules13081875
Chicago/Turabian StyleSzabó, László F. 2008. "Rigorous Biogenetic Network for a Group of Indole Alkaloids Derived from Strictosidine" Molecules 13, no. 8: 1875-1896. https://doi.org/10.3390/molecules13081875
APA StyleSzabó, L. F. (2008). Rigorous Biogenetic Network for a Group of Indole Alkaloids Derived from Strictosidine. Molecules, 13(8), 1875-1896. https://doi.org/10.3390/molecules13081875