Synthesis, IR Spectra, Crystal Structure and DFT Studies on 1-Acetyl-3-(4-Chlorophenyl)-5-(4-Methylphenyl)-2-Pyrazoline

Abstract
:Introduction
Results and Discussion
Description of the crystal structure


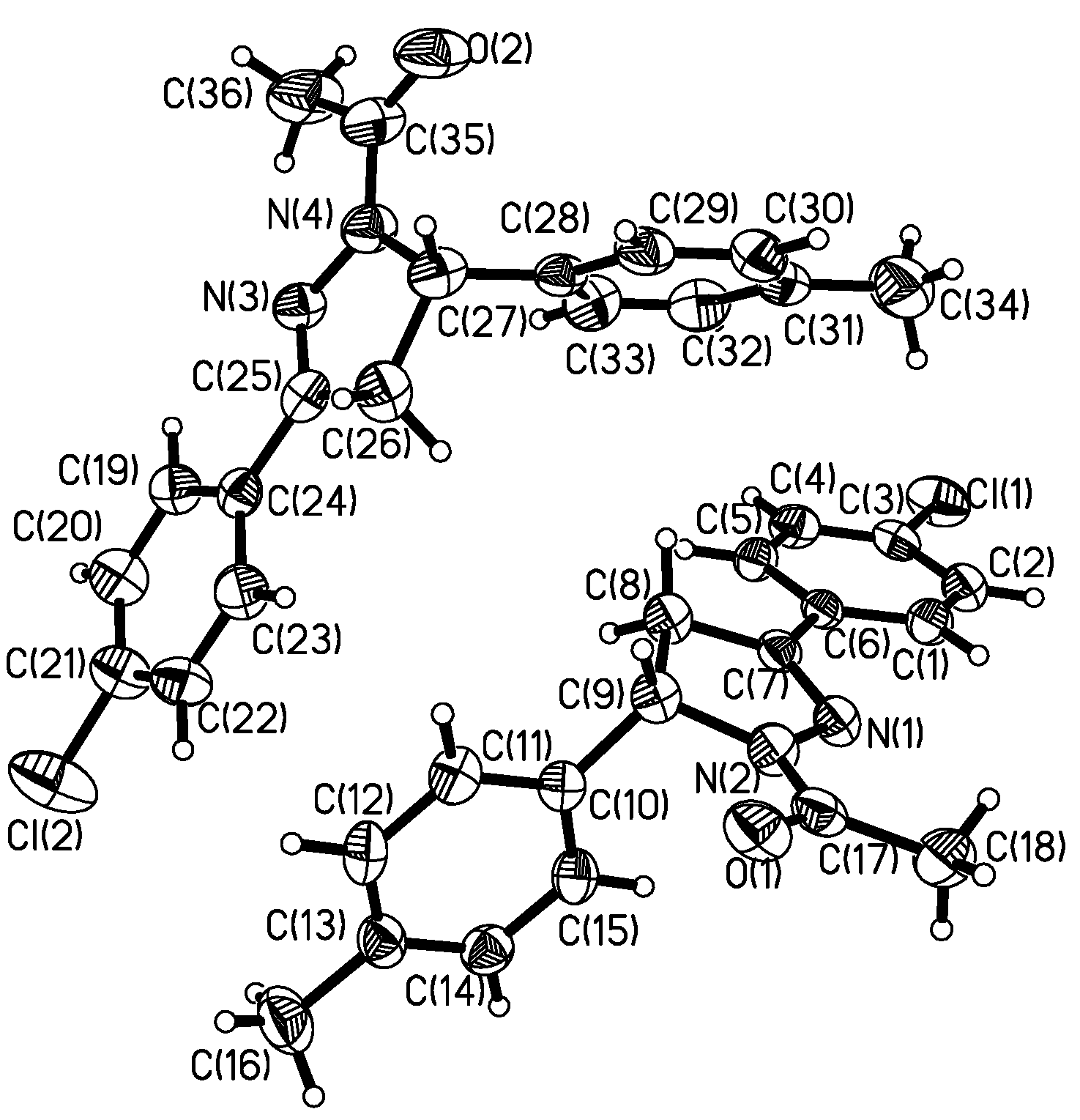{kind=link}
{kind=link}
{kind=link}
| Bond lengths (Å) | Exp. | Bond lengths | Exp. | B3LYP/6-311G** |
|---|---|---|---|---|
| Cl(1)-C(3) | 1.745(4) | Cl(2)-C(21) | 1.725(5) | 1.7577 |
| O(1)-C(17) | 1.220(4) | O(2)-C(35) | 1.224(5) | 1.2171 |
| N(1)-C(7) | 1.293(4) | N(3)-C(25) | 1.292(4) | 1.2889 |
| N(1)-N(2) | 1.395(4) | N(3)-N(4) | 1.398(4) | 1.3699 |
| N(2)-C(17) | 1.372(5) | N(4)-C(35) | 1.363(5) | 1.3826 |
| N(2)-C(9) | 1.483(4) | N(4)-C(27) | 1.488(5) | 1.4863 |
| C(1)-C(2) | 1.382(5) | C(19)-C(20) | 1.378(5) | 1.3857 |
| C(5)-C(6) | 1.390(5) | C(23)-C(24) | 1.395(5) | 1.4018 |
| C(6)-C(7) | 1.471(5) | C(24)-C(25) | 1.475(5) | 1.4639 |
| C(8)-C(9) | 1.551(5) | C(26)-C(27) | 1.541(5) | 1.5523 |
| C(9)-C(10) | 1.515(5) | C(27)-C(28) | 1.510(5) | 1.5165 |
| C(10)-C(15) | 1.375(5) | C(28)-C(29) | 1.374(5) | 1.3933 |
| C(10)-C(11) | 1.380(5) | C(28)-C(33) | 1.388(5) | 1.3987 |
| C(13)-C(16) | 1.521(5) | C(31)-C(34) | 1.522(6) | 1.5095 |
| C(17)-C(18) | 1.502(5) | C(35)-C(36) | 1.495(6) | 1.513 |
| Bond angles (°) | Exp. | Bond angles (°) | Exp. | B3LYP/6-311G** |
| C(7)-N(1)-N(2) | 108.3(3) | C(25)-N(3)-N(4) | 107.4(3) | 109.3937 |
| N(1)-N(2)-C(9) | 113.3(3) | N(3)-N(4)-C(27) | 113.3(3) | 113.5694 |
| N(1)-C(7)-C(8) | 113.8(3) | N(3)-C(25)-C(26) | 114.7(4) | 113.0852 |
| C(7)-C(8)-C(9) | 103.0(3) | C(25)-C(26)-C(27) | 102.8(3) | 102.7091 |
| N(2)-C(9)-C(8) | 100.9(3) | N(4)-C(27)-C(26) | 101.1(3) | 100.7838 |
| C(17)-N(2)-N(1) | 122.9(3) | C(35)-N(4)-N(3) | 122.8(4) | 122.7854 |
| C(2)-C(1)-C(6) | 121.4(4) | C(20)-C(19)-C(24) | 120.6(4) | 120.984 |
| C(3)-C(4)-C(5) | 119.1(4) | C(23)-C(22)-C(21) | 120.2(4) | 119.1608 |
| C(1)-C(6)-C(7) | 121.1(4) | C(19)-C(24)-C(25) | 121.0(4) | 120.9516 |
| C(15)-C(10)-C(11) | 117.2(3) | C(29)-C(28)-C(33) | 118.3(4) | 118.401 |
| C(13)-C(14)-C(15) | 121.8(4) | C(31)-C(32)-C(33) | 121.6(4) | 121.1049 |
| C(12)-C(13)-C(16) | 121.1(4) | C(32)-C(31)-C(34) | 121.8(5) | 120.8844 |
| O(1)-C(17)-N(2) | 119.5(4) | O(2)-C(35)-N(4) | 119.3(5) | 119.787 |
| O(1)-C(17)-C(18) | 124.4(4) | O(2)-C(35)-C(36) | 124.0(5) | 123.9266 |
| N(2)-C(17)-C(18) | 116.1(4) | N(4)-C(35)-C(36) | 116.8(4) | 116.2863 |
Optimized geometry

Vibrational frequency

| Assignments | Exp. IR (with KBr) | Calculated ( B3LYP/6-311G** ) |
|---|---|---|
| phenyl ring C-H str. | 3066 | 3080-3030 |
| acetyl C-H str. | 3033 | 3026 |
| pyrazolinyl ring C-H str. | 2969 | 2966 |
| methyl group C-H str. | 2885 | 2901 |
| C=O str. | 1666 | 1681 |
| phenyl ring C=C str.+ C=N str. | 1591 | 1591-1577 |
| phenyl ring C=C str. | 1507 | 1486 |
| methyl group C-H bend | 1430 | 1437 |
| phenyl ring C-H bend + pyrazolinyl ring C-H bend | 1319 | 1328 |
| pyrazolinyl ring C-H bend + N-N str. | 1248 | 1248 |
| pyrazolinyl ring C-H bend + N-N str. | 1144 | 1138 |
| pyrazolinyl ring C-H bend | 1089 | 1088 |
| methyl group C-H bend | 1014 | 1019-1011 |
| phenyl ring C-H bend | 953 | 950 |
| phenyl ring C-H twist. | 819 | 815 |
| skeleton deformation + C-Cl str. | 726 | 715 |
| skeleton deformation | 627 | 630 |
Thermodynamic properties
| T (K) | C0p,m (J·mol-1·K-1) | S0m (J·mol-1·K-1) | H0m (kJ·mol-1) |
|---|---|---|---|
| 100.0 | 147.83 | 422.98 | 9.79 |
| 200.0 | 239.08 | 552.87 | 29.05 |
| 298.1 | 337.74 | 666.57 | 57.32 |
| 300.0 | 339.61 | 668.66 | 57.95 |
| 400.0 | 435.62 | 779.80 | 96.81 |
| 500.0 | 516.58 | 886.01 | 144.56 |
| 600.0 | 581.88 | 986.18 | 199.60 |
| 700.0 | 634.50 | 1079.97 | 260.51 |
| 800.0 | 677.47 | 1167.59 | 326.18 |
Experimental
General
Synthesis
Theoretical methods
Crystal structure determination
| Empirical formula | C36H34Cl2 N4O2 |
|---|---|
| Formula weight | 625.57 |
| Temperature | 293(2) K |
| Wavelength | 0.71073 Å |
| Crystal system, space group | Monoclinic, P2(1)/c |
| Unit cell dimensions | a = 18.158(16) Å |
| b = 13.461(12) Å β= 112.654(16) o | |
| c = 14.751(14) Å | |
| Volume | 3327(5) Å3 |
| Z, Calculated density | 4, 1.249 Mg/m3 |
| Absorption coefficient | 0.233 |
| F(000) | 1312 |
| θ range for data collection | 1.94 to 25.02 ° |
| Limiting indices | -21 ≤ h ≤ 19, -13 ≤ k ≤ 16, -17 ≤ l ≤15 |
| Reflections collected / unique | 16668 / 5851 [Rin t= 0.0835] |
| Refinement method | Full-matrix least-squares on F2 |
| Data / restraints / parameters | 5851 / 18 / 401 |
| Goodness-of-fit on F2 | 1.001 |
| Final R indices [I>2σ(I)] | R1 = 0.0500, wR2 = 0.1123 |
| R indices (all data) | R1 = 0.1730, wR2 = 0.1586 |
| Largest diff. peak and hole | 0.180 and -0.227 e. Å-3 |
Acknowledgements
References
- Mason, W. T. Fluorescent and Luminescent Probes for Biological Activity: A Practical Guide to Technology for Quantitative Real-time Analysis; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Takahashi, A.; Camacho, P.; Lechleiter, J. D.; Herman, B. Measurement of Intracellular Calcium. Physiol. Rev. 1999, 79, 1089–1125. [Google Scholar]
- Burdette, S. C.; Walkup, G.. K.; Spingler, B.; Tsien, R.Y.; Lippard, S. J. Fluorescent Sensors for Zn2+ Based on a Fluorescein Platform: Synthesis, Properties and Intracellular Distribution. J. Am. Chem. Soc. 2001, 123, 7831–7841. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Kikuchi, K.; Urano, Y.; Nagano, T. Improvement and Biological Applications of Fluorescent Probes for Zinc, ZnAFs. J. Am. Chem. Soc. 2002, 124, 6555–6562. [Google Scholar] [CrossRef]
- Espósito, B. P.; Epsztejn, S.; Breuer, W.; Cabantchik, Z. I. A Review of Fluorescence Methods for Assessing Labile Iron in Cells and Biological Fluids. Anal. Biochem. 2002, 304, 1–18. [Google Scholar]
- Takahashi, A.; Zhang, Y. P.; Centonze, V. E.; Herman, B. Measurement of Mitochondrial pH In Situ. Biotechniques 2001, 30, 804–815. [Google Scholar]
- Rivett, D. E.; Rosevear, J.; Wilshire, J. F. K. The preparation and spectroscopic properties of some di- and tri-substituted 1, 3, 5-triphenyl-2-pyrazolines and related 2-pyrazolines. Aust. J. Chem. 1983, 36, 1649–1658. [Google Scholar] [CrossRef]
- de Silva, A. P.; Nimal Gunaratne, H. Q.; Gunnlaugsson, T.; Nieuwenhuizen, M. Fluorescent switches with high selectivity towards sodium ions: correlation of ion-induced conformation switching with fluorescence function. Chem. Commun. 1996, 1967–1968. [Google Scholar]
- Rurack, K.; Resch-Genger, U.; Spieles, M.; J.L., Bricks. Cation-triggered switching on of the red/near infra-red (NIR) fluorescence of rigid fluorophore-spacer-receptor ionophores. Chem. Commun. 2000, 2103–2104. [Google Scholar]
- Rurack, K.; Bricks, J. L.; Schulz, B.; Maus, M.; Reck, G.; Resch-Genger, U. Substituted 1,5-Diphenyl-3-benzothiazol-2-yl-Ĕ2-pyrazolines: Synthesis, X-ray Structure, Photophysics, and Cation Complexation Properties. J. Phys. Chem. A. 2000, 104, 6171–6188. [Google Scholar] [CrossRef]
- Fahrni, C. J.; Yang, L. C.; VanDerveer, D. G. Tuning the Photoinduced Electron-Transfer Thermodynamics in 1,3,5-Triaryl-2-pyrazoline Fluorophores: X-ray Structures, Photophysical Characterization, Computational Analysis, and in Vivo Evaluation. J. Am. Chem. Soc. 2003, 125, 3799–3812. [Google Scholar] [CrossRef]
- Wagner, A.; Schellhammer, C. W.; Petersen, S. Aryl-Ĕ2-pyrazolines as Optical Brighteners. Angew. Chem. Int. Ed. Engl. 1966, 5, 699–704. [Google Scholar] [CrossRef]
- Labanowski, J. K.; Andzelm, J. Density Functional Methods in Chemistry; Springer-Verlag: New York, USA, 1991. [Google Scholar]
- Oliphant, N.; Bartlett, R. J. A systematic comparison of molecular properties obtained using Hartree-Fock, a hybrid Hartree-Fock density-functional-theory, and coupled-cluster methods. J. Chem. Phys. 1994, 100, 6550–6556. [Google Scholar] [CrossRef]
- Dickson, R. M.; Becke, A. D. Basis-set-free local density-functional calculations of geometries of polyatomic molecules. J. Chem. Phys. 1993, 99, 3898–3902. [Google Scholar] [CrossRef]
- Johnson, B. G.; Gill, P. M. W.; Pople, J. A. The performance of a family of density functional methods. J. Chem. Phys. 1993, 98, 5612–5618. [Google Scholar] [CrossRef]
- Yakuphanoglu, F.; Atalay, Y.; Sekerci, M. A theoretical study on N-phenyl-N′-(2-thienyl-methylene) hydrazine. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2007, 66, 438–441. [Google Scholar] [CrossRef]
- Jian, F. F.; Zhao, P. S.; Guo, H. M.; Li, Y. F. Synthesis, characterization, crystal structure and DFT studies on 1-acetyl-3-(2,4-dichloro-5-fluoro-phenyl)-5-phenyl-pyrazoline. Spectrochim. Acta. A Mol. Biomol. Spectrosc. 2008, 69, 647–653. [Google Scholar] [CrossRef]
- Jeffrey, G. A.; Maluszynska, H.; Mitra, J. Hydrogen bonding in nucleosides and nucleotides. Int. J. Biol. Macromol. 1985, 7, 336–348. [Google Scholar] [CrossRef]
- Jian, F. F.; Zhao, P. S.; Yu, Q.; Wang, Q. X.; Jiao, K. Density Functional Calculations, Synthesis, and Characterization on Two Novel Quadruple Hydrogen-Bonded Supramolecular Complexes. J. Phys. Chem. A. 2004, 108, 5258–5267. [Google Scholar] [CrossRef]
- Frish, A.; Nielsen, A. B.; Holder, A. J. Gaussview Users Manual; Gaussian Inc.: Pittsburgh, PA, USA, 2000. [Google Scholar]
- Peng, C.; Ayala, P. Y.; Schlegel, H. B.; Frisch, M. J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 1996, 17, 49–56. [Google Scholar] [CrossRef]
- risch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03.; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Sheldrick, G. M. SHELXTL, v5 Reference Manual, Siemens Analytical X-Ray Systems; Bruker AXS, Inc: Madison, WI, USA, 1997. [Google Scholar]
- Wilson, A. J. International Table for X-Ray Crystallography; Kluwer Academic: Dordrecht, The Netherlands, 1992; Volume C; pp. 500-502 and pp. 219-222. [Google Scholar]
- Sample Availability: Samples of the title compound are available from the authors.
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guo, H.-M.; Wang, L.-T.; Zhang, J.; Zhao, P.-S.; Jian, F.-F. Synthesis, IR Spectra, Crystal Structure and DFT Studies on 1-Acetyl-3-(4-Chlorophenyl)-5-(4-Methylphenyl)-2-Pyrazoline. Molecules 2008, 13, 2039-2048. https://doi.org/10.3390/molecules13092039
Guo H-M, Wang L-T, Zhang J, Zhao P-S, Jian F-F. Synthesis, IR Spectra, Crystal Structure and DFT Studies on 1-Acetyl-3-(4-Chlorophenyl)-5-(4-Methylphenyl)-2-Pyrazoline. Molecules. 2008; 13(9):2039-2048. https://doi.org/10.3390/molecules13092039
Chicago/Turabian StyleGuo, Huan-Mei, Lin-Tong Wang, Jing Zhang, Pu-Su Zhao, and Fang-Fang Jian. 2008. "Synthesis, IR Spectra, Crystal Structure and DFT Studies on 1-Acetyl-3-(4-Chlorophenyl)-5-(4-Methylphenyl)-2-Pyrazoline" Molecules 13, no. 9: 2039-2048. https://doi.org/10.3390/molecules13092039
APA StyleGuo, H. -M., Wang, L. -T., Zhang, J., Zhao, P. -S., & Jian, F. -F. (2008). Synthesis, IR Spectra, Crystal Structure and DFT Studies on 1-Acetyl-3-(4-Chlorophenyl)-5-(4-Methylphenyl)-2-Pyrazoline. Molecules, 13(9), 2039-2048. https://doi.org/10.3390/molecules13092039