In Vitro and In Vivo Evaluation of Microparticulate Drug Delivery Systems Composed of Macromolecular Prodrugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Macromolecular prodrugs of MMC and their microparticulate formulations
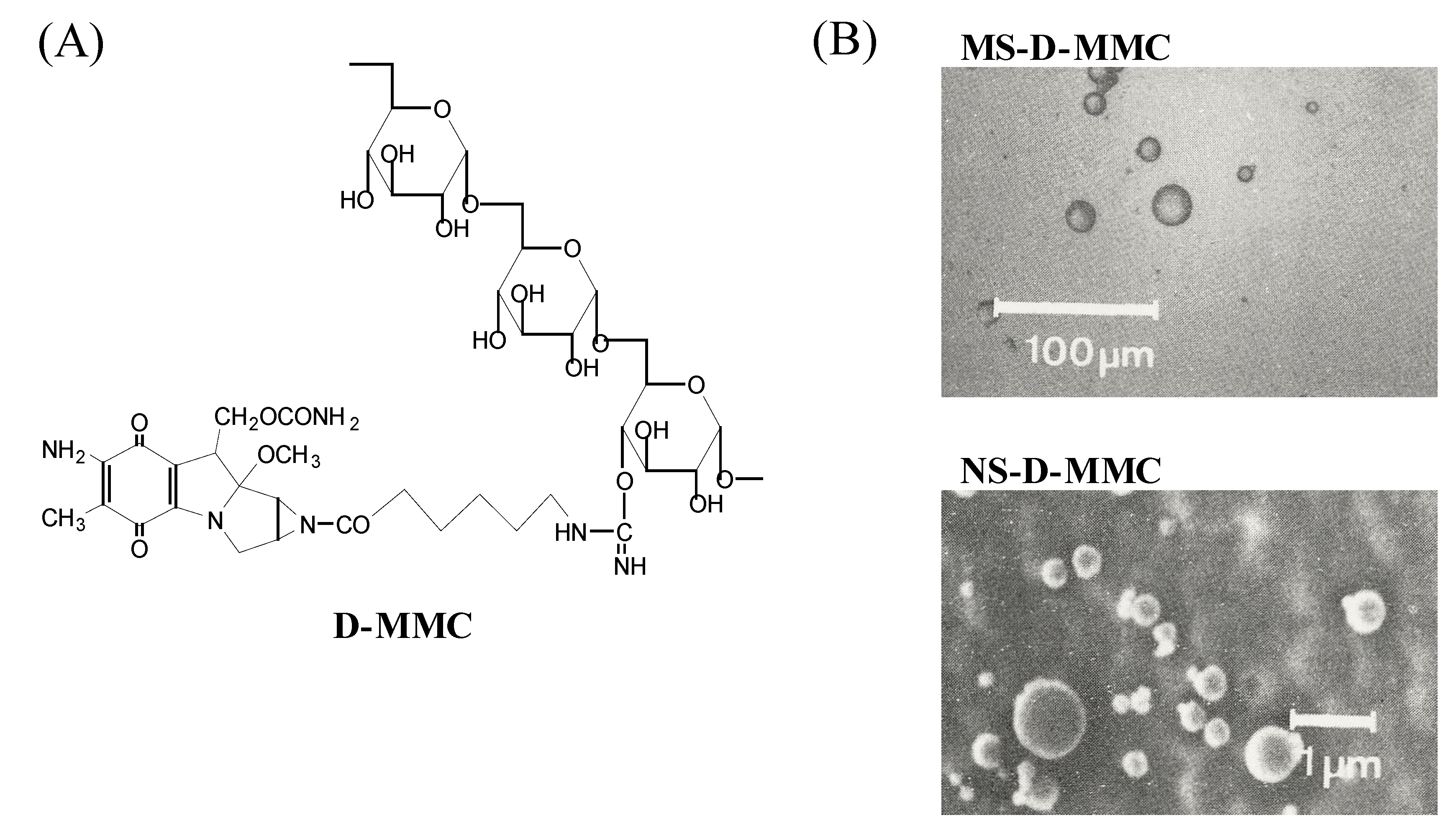
2.1. Gelatin nano- and microspheres containing dextran-MMC conjugate (D-MMC)

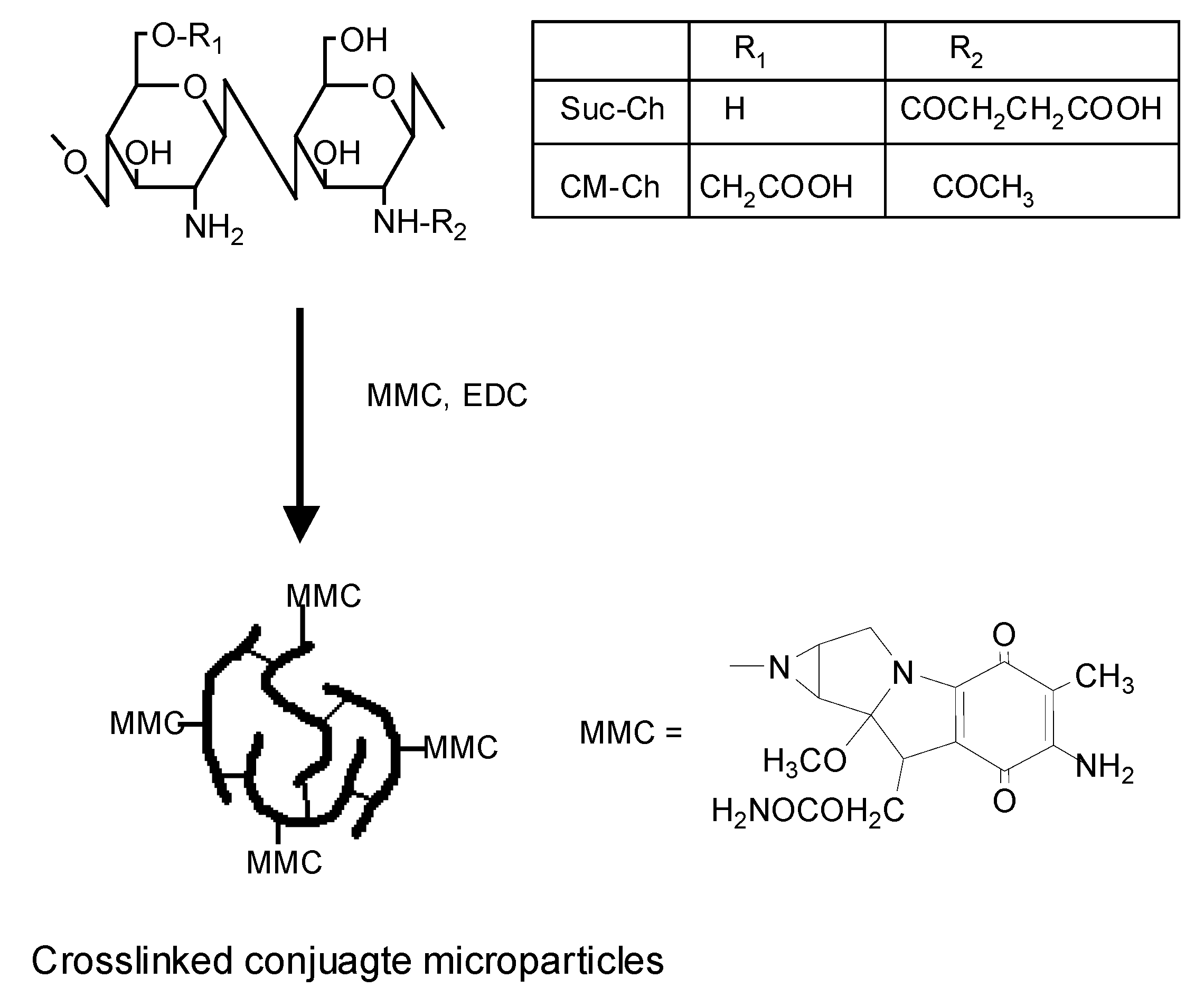
2.2. N-Succinyl-chitosan-MMC and 6-O-carboxymethylchitin-MMC conjugate microparicles

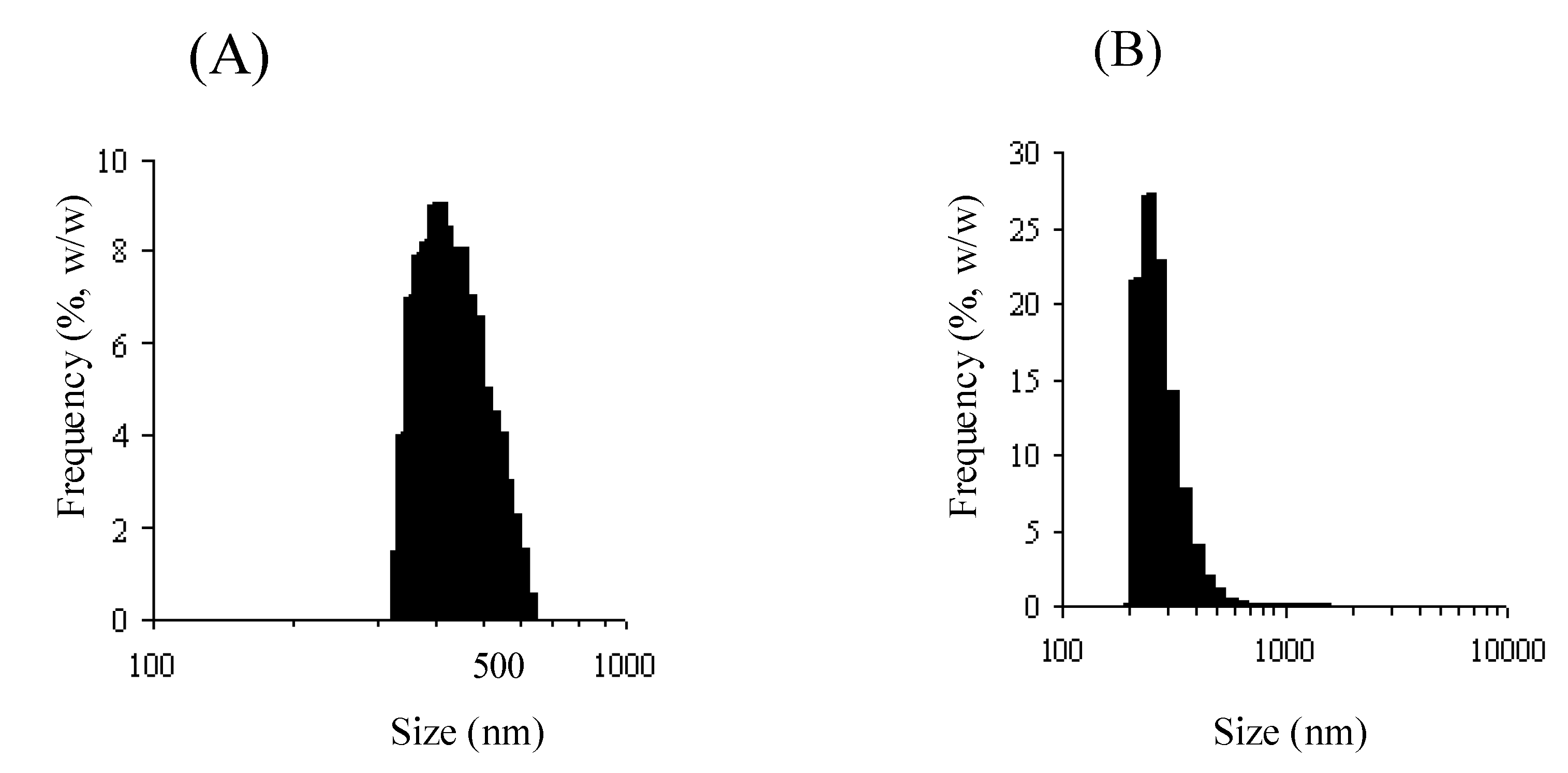
2.3. N-Succinyl-chitosan-MMC and 6-O-carboxymethylchitin-MMC conjugate nanoparticles

3. Conjugates of drugs with poly(D,L-lactic-co-glycolic acid) and their micro- and nanoparticulate dosage forms
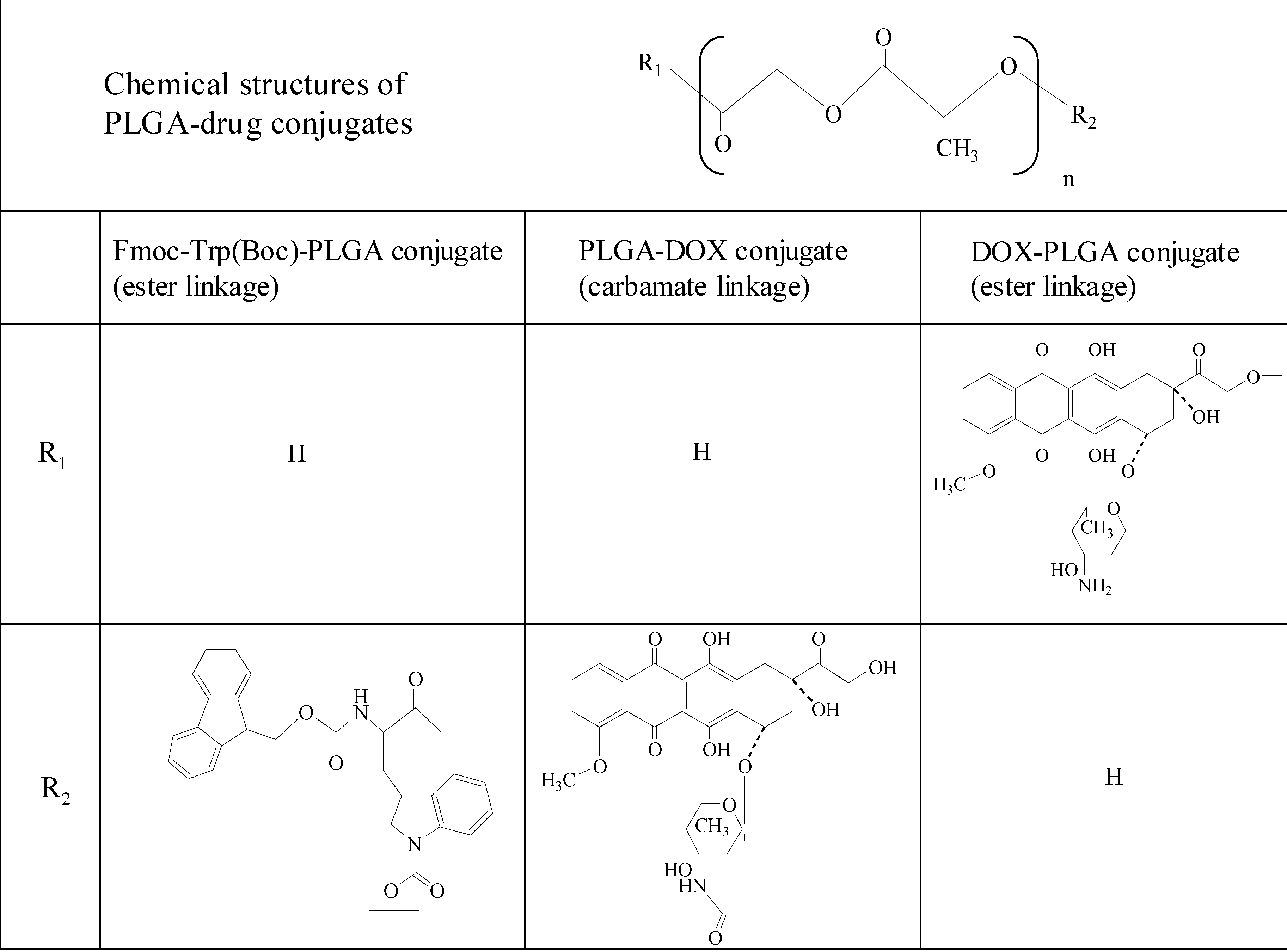
3.1. Poly(D,L-lactic-co-glycolic acid) (PLGA)-drug conjugate microspheres
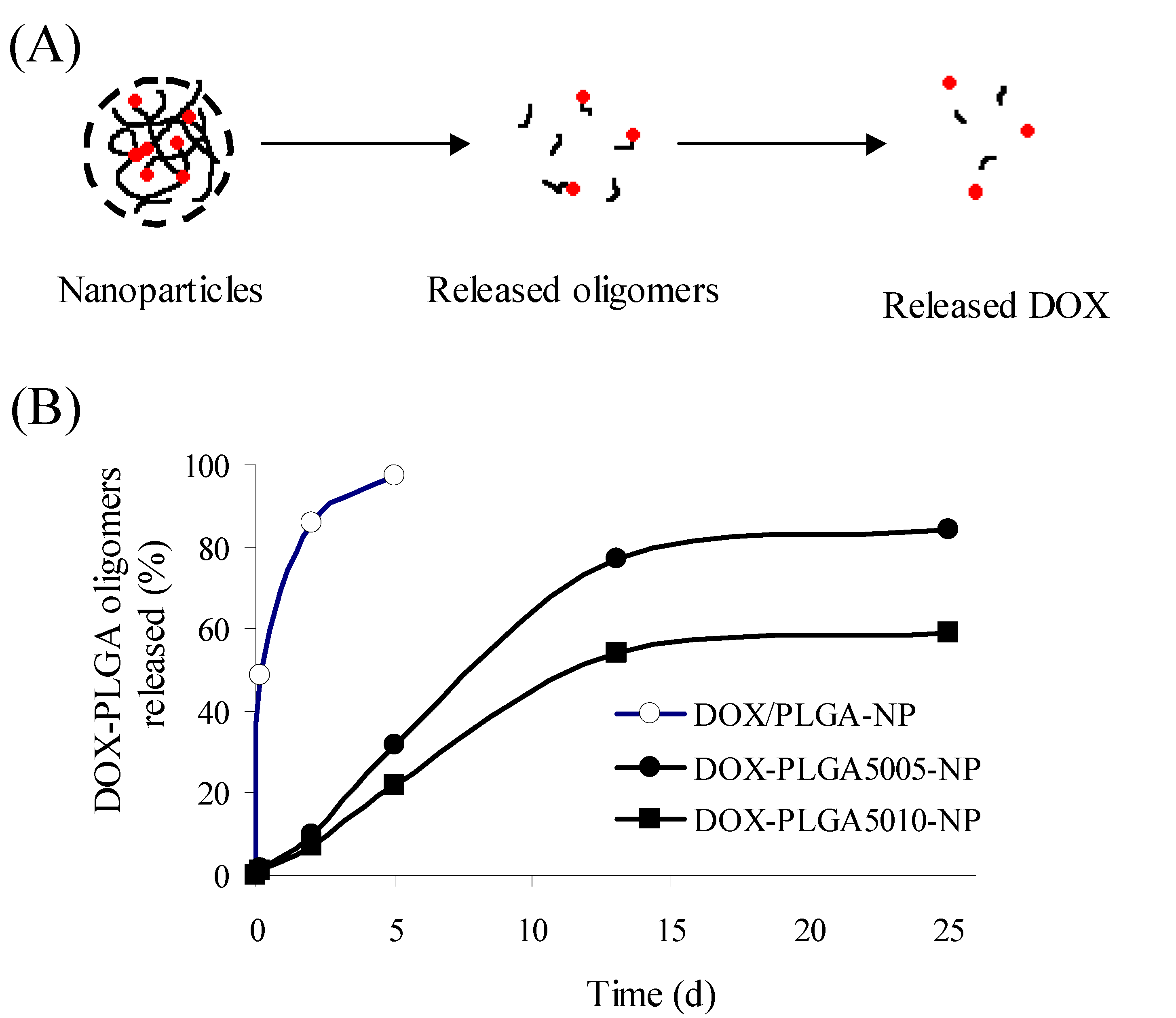
3.2. PLGA-doxorubicin conjugate nanoparticles


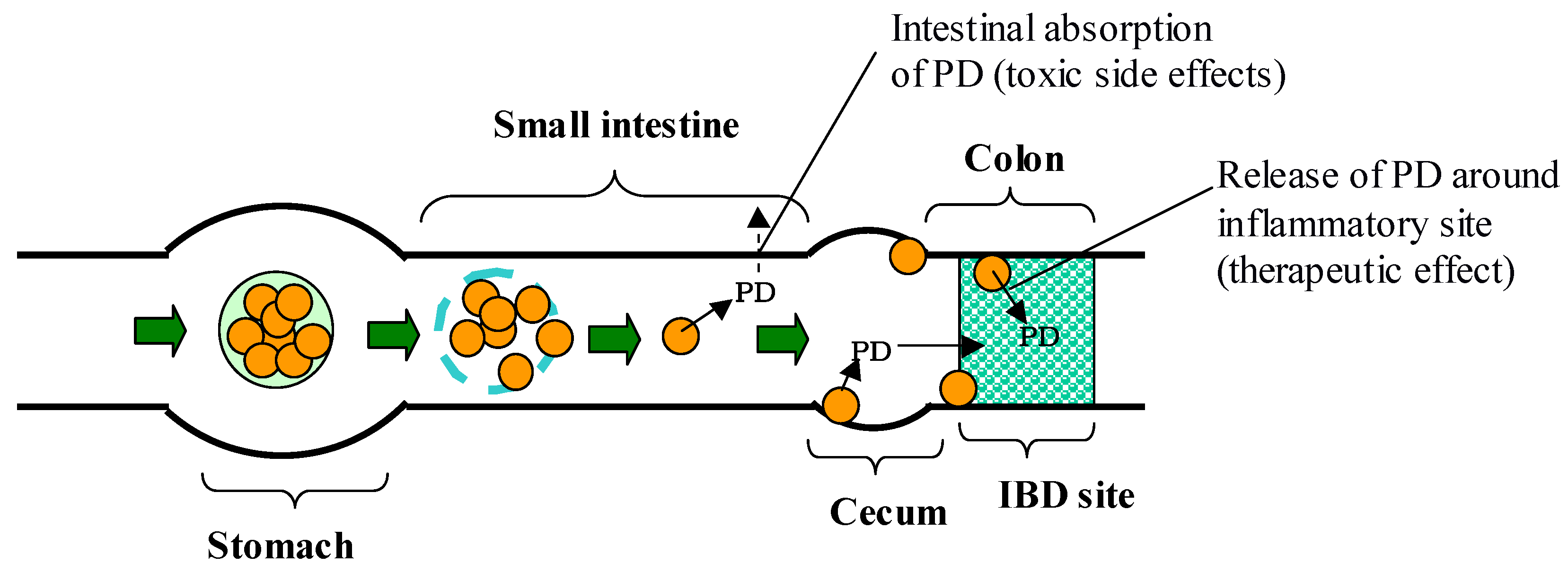
4. Microspheres composed of chitosan-succinyl-prednisolone conjugate and their enteric-coated microparticles


5. Conclusions
Abbreviations
| Ch | chitosan |
| Ch-SP | Chitosan-prednisolone conjugate |
| Ch-SP-MS | Ch-SP microspheres |
| Ch-SP-MS/EuL | Eudragit L100-coated Ch-SP-MS |
| Ch-SP-MS/EuS | Eudragit L100-coated Ch-SP-MS |
| CM-Ch | 6-O-Carboxymethylchitin |
| CM-Dextran | Carboxymethyl-dextran |
| CM-Ch-MMC | CM-Ch-mitomycin C conjugtate |
| CM-Ch-MMC-NP | CM-Ch-MMC nanoparticles |
| CPT | Camptothecin |
| D-MMC | Dextran-Mitomycin C conjugate |
| DOX | Doxorubicin |
| DOX-PLGA-NP | DOX-poly(D,L-lactic-co-glycolic acid) nanoparticles |
| DOX/PLGA-NP | DOX-poly(D,L-lactic-co-glycolic acid) nanoparticles mixture |
| EDC | 1-Ethyl-3-(3-dimehylaminopropyl)carbodiimide hydrochloride |
| EPR | Enhanced permeability retention |
| Fmoc-Trp(Boc) | N-(9-Fluorenylmethoxycarbonyl)-N-tert-butoxycarbonyl-L-tryptophan |
| IBD | Inflammatory bowel disease |
| MMC | Mitomycin C |
| MPO | Myeloperoxidase |
| MS | Microspheres |
| MS-D-MMC | D-MMC-loaded microspheres |
| MS-MMC | MMC-containing micropsheres |
| NS | Nanospheres |
| NS-D-MMC | D-MMC-loaded nanospheres |
| NS-MMC | MMC-containing nanospheres |
| PTX | Paclitaxel |
| PLA | Poly(D,L-lactic acid) |
| PLGA | Poly(D,L-lactic-co-glycolic acid) |
| RES | Reticuloendothelial system |
| SP | Succinyl-prednisolone |
| Suc-Ch | N-Succinyl-chitosan |
| Suc-Ch-MMC | Suc-Ch-MMC conjugate |
| Suc-Ch-MMC-NP | Suc-Ch-MMC nanoparticles |
| TNBS | 2,4,6-Trinitro-benzenesulfonic acid |
References
- Cavallaro, G.; Pitarresi, G.; Licciardi, M.; Giammona, G. Polymeric prodrug for release of an antitumoral agent by specific enzymes. Bioconjug Chem. 2001, 12, 143–151. [Google Scholar] [CrossRef]
- Kratz, F.; Abu Ajaj, K.; Warnecke, A. Anticancer carrier-linked prodrugs in clinical trials. Expert Opin. Investig. Drugs 2007, 16, 1037–1058. [Google Scholar]
- Julyan, P.J.; Seymour, L.W.; Ferry, D.R.; Daryani, S.; Boivin, C.M.; Doran, J.; David, M.; Anderson, D.; Christodoulou, C.; Young, A.M.; Hesslewood, S.; Kerr, D.J. Preliminary clinical study of the distribution of HPMA copolymers bearing doxorubicin and galactosamine. J. Control. Release 1999, 57, 281–290. [Google Scholar] [CrossRef]
- Ulbrich, K.; Etrych, T.; Chytil, P.; Jelínková, M.; Ríhová, B. HPMA copolymers with pH-controlled release of doxorubicin: in vitro cytotoxicity and in vivo antitumor activity. J. Control. Release 2003, 87, 33–47. [Google Scholar] [CrossRef]
- Singer, J.W.; Bhatt, R.; Tulinsky, J.; Buhler, K.R.; Heasley, E.; Klein, P.; de Vries, P. Water-soluble poly-(L-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J. Control. Release 2001, 74, 243–247. [Google Scholar] [CrossRef]
- Zou, Y.; Fu, H.; Ghosh, S.; Farquhar, D.; Klostergaard, J. Antitumor activity of hydrophilic Paclitaxel copolymer prodrug using locoregional delivery in human orthotopic non-small cell lung cancer xenograft models. Clin. Cancer Res. 2004, 10, 7382–7391. [Google Scholar] [CrossRef]
- Takakura, Y.; Matsumoto, S.; Hashida, M.; Sezaki, H. Enhanced lymphatic delivery of mitomycin C conjugated with dextran. Cancer Res. 1984, 44, 2505–2510. [Google Scholar]
- Okuno, S.; Harada, M.; Yano, T.; Yano, S.; Kiuchi, S.; Tsuda, N.; Sakamura, Y.; Imai, J.; Kawaguchi, T.; Tsujihara, K. Complete regression of xenografted human carcinomas by camptothecin analogue-carboxymethyl dextran conjugate (T-0128). Cancer Res. 2000, 60, 2988–2995. [Google Scholar]
- Trouet, A.; Masquelier, M.; Baurain, R.; Deprez-De Campeneere, D. A covalent linkage between daunorubicin and proteins that is stable in serum and reversible by lysosomal hydrolases, as required for a lysosomotropic drug-carrier conjugate: in vitro and in vivo studies. Natl. Acad. Sci. U S A. 1982, 79, 626–629. [Google Scholar]
- Kaneo, Y.; Tanaka, T.; Iguchi, S. Preparation and properties of a mitomycin C-albumin conjugate. Chem. Pharm. Bull. 1990, 38, 2614–2616. [Google Scholar] [CrossRef]
- Takakura, Y.; Takagi, A.; Hashida, M.; Sezaki, H. Disposition and tumor localization of mitomycin C-dextran conjugates in mice. Pharm. Res. 1987, 4, 293–300. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kuroda, M.; Tabata, Y.; Ikada, Y. Body distribution of dextran derivatives with electric charges after intravenous administration. Int. J. Pharm. 1995, 113, 149–157. [Google Scholar]
- Takakura, Y.; Fujita, T.; Hashida, M.; Sezaki, H. Disposition characteristics of macromolecules in tumor-bearing mice. Pharm. Res. 1990, 7, 339–346. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Unezaki, S.; Maruyama, K.; Ishida, O.; Suginaka, A.; Hosoda, J.; Iwatsuru, M. Enhanced tumor targeting and improved antitumor activity of doxorubicin by long-circulating liposomes containing amphipathic poly(ethylene glycol). Int. J. Pharm. 1995, 126, 41–48. [Google Scholar] [CrossRef]
- Nakanishi, T.; Fukushima, S.; Okamoto, K.; Suzuki, M.; Matsumura, Y.; Yokoyama, M.; Okano, T.; Sakurai, Y.; Kataoka, K. Development of the polymer micelle carrier system for doxorubicin. J. Control. Release 2001, 74, 295–302. [Google Scholar] [CrossRef]
- Kato, A.; Takakura, Y.; Hashida, M.; Kimura, T.; Sezaki, H. Physico-chemical and antitumor characteristics of high molecular weight prodrugs of mitomycin C. Chem. Pharm. Bull. 1982, 30, 2951–2957. [Google Scholar] [CrossRef]
- Duncan, R.; Hume, I.C.; Kopecková, P.; Ulbrich, K.; Strohalm, J.; Kopecek, J. Anticancer agents coupled to N-(2-hydroxypropyl)methacrylamide copolymers. 3. Evaluation of daunomycin conjugates against mouse leukaemia L1210 in vivo. J. Control. Release 1989, 10, 51–63. [Google Scholar] [CrossRef]
- Pithayanukul, P.; Onishi, H.; Nagai, T. In vitro pH-dependent drug release from N4-(4-carboxybutyryl)-1-beta-D-arabinofuranosylcytosine and its conjugate with poly-L-lysine or decylenediamine-dextran T70. Chem. Pharm. Bull. 1989, 37, 1587–1590. [Google Scholar] [CrossRef]
- Kovár, M.; Kovár, L.; Subr, V.; Etrych, T.; Ulbrich, K.; Mrkvan, T.; Loucká, J.; Ríhová, B. HPMA copolymers containing doxorubicin bound by a proteolytically or hydrolytically cleavable bond: comparison of biological properties in vitro. J. Control. Release 2004, 99, 301–314. [Google Scholar] [CrossRef]
- Oyama, T.; Kawamura, M.; Abiko, T.; Izumi, Y.; Watanabe, M.; Kumazawa, E.; Kuga, H.; Shiose, Y.; Kobayashi, K. Hyperthermia-enhanced tumor accumulation and antitumor efficacy of a doxorubicin-conjugate with a novel macromolecular carrier system in mice with non-small cell lung cancer. Oncol. Rep. 2007, 17, 653–659. [Google Scholar]
- Conover, C.D.; Greenwald, R.B.; Pendri, A.; Gilbert, C.W.; Shum, K.L. Camptothecin delivery systems: enhanced efficacy and tumor accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother. Pharmacol. 1998, 42, 407–414. [Google Scholar] [CrossRef]
- Caiolfa, V.R.; Zamai, M.; Fiorino, A.; Frigerio, E.; Pellizzoni, C.; d'Argy, R.; Ghiglieri, A.; Castelli, M.G.; Farao, M.; Pesenti, E.; Gigli, M.; Angelucci, F.; Suarato, A. Polymer-bound camptothecin: initial biodistribution and antitumour activity studies. J. Control. Release 2000, 65, 105–119. [Google Scholar] [CrossRef]
- Harada, M.; Sakakibara, H.; Yano, T.; Suzuki, T.; Okuno, S. Determinants for the drug release from T-0128, camptothecin analogue-carboxymethyl dextran conjugate. J. Control. Release 2000, 69, 399–412. [Google Scholar] [CrossRef]
- Sugahara, S.; Kajiki, M.; Kuriyama, H.; Kobayashi, T.R. Complete regression of xenografted human carcinomas by a paclitaxel-carboxymethyl dextran conjugate (AZ10992). J. Control. Release 2007, 117, 40–50. [Google Scholar] [CrossRef]
- Kratz, F.; Abu Ajaj, K.; Warnecke, A. Anticancer carrier-linked prodrugs in clinical trials. Expert Opin. Investig. Drugs. 2007, 16, 1037–1058. [Google Scholar] [CrossRef]
- Endo, N.; Kato, Y.; Takeda, Y.; Saito, M.; Umemoto, N.; Kishida, K.; Hara, T. In vitro cytotoxicity of a human serum albumin-mediated conjugate of methotrexate with anti-MM46 monoclonal antibody. Cancer Res. 1987, 47, 1076–1080. [Google Scholar]
- Burger, A.M.; Hartung, G.; Stehle, G.; Sinn, H.; Fiebig, H.H. Pre-clinical evaluation of a methotrexate-albumin conjugate (MTX-HSA) in human tumor xenografts in vivo. Int. J. Cancer 2001, 92, 718–724. [Google Scholar] [CrossRef]
- Kato, Y.; Saito, M.; Fukushima, H.; Takeda, Y.; Hara, T. Antitumor activity of 1-beta-D-arabinofuranosylcytosine conjugated with polyglutamic acid and its derivative. Cancer Res. 1984, 44, 25–30. [Google Scholar]
- Onishi, H.; Pithayanukul, P.; Nagai, T. Antitumor characteristics of the conjugate of N4-(4-carboxybutyryl)-ara-C with ethylenediamine-introduced dextran and its resistance to cytidine deaminase. Drug Des. Deliv. 1990, 6, 273–280. [Google Scholar]
- Tanaka, T.; Kaneo, Y.; Iguchi, S. Properties of mitomycin C-albumin conjugates in vitro and in vivo. Bioconjug. Chem. 1991, 2, 261–269. [Google Scholar] [CrossRef]
- Kato, Y.; Onishi, H.; Machida, Y. Efficacy of lactosaminated and intact N-succinylchitosan-mitomycin C conjugates against M5076 liver metastatic cancer. J. Pharm. Pharmacol. 2002, 54, 529–537. [Google Scholar] [CrossRef]
- Sugibayashi, K.; Morimoto, Y.; Nadai, T.; Kato, Y.; Hasegawa, A.; Arita, T. Drug-carrier property of albumin microspheres in chemotherapy. II. Preparation and tissue distribution in mice of microsphere-entrapped 5-fluorouracil. Chem. Pharm. Bull. 1979, 27, 204–209. [Google Scholar] [CrossRef]
- Kanke, M.; Simmons, G.H.; Weiss, D.L.; Bivins, B.A.; DeLuca, P.P. Clearance of 141Ce-labeled microspheres from blood and distribution in specific organs following intravenous and intraarterial administration in beagle dogs. J. Pharm. Sci. 1980, 69, 755–762. [Google Scholar] [CrossRef]
- Bazile, D.; Prud’homme, C.; Bassoullet, M.T.; Marlard, M.; Spenlehauer, G.; Veillard, M. Stealth PEG-PLA nanoparticles avoid uptake by the mononuclear phagocytes system. J. Pharm. Sci. 1995, 84, 493–498. [Google Scholar] [CrossRef]
- Desai, M.P.; Labhasetwar, V.; Amidon, G.L.; Levy, R.J. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm. Res. 1996, 13, 1838–1845. [Google Scholar] [CrossRef]
- Dunn, S.E.; Coombes, A.G.A.; Garnett, M.C.; Davis, S.S.; Davies, M.C.; Illum, L. In vitro cell interaction and in vivo biodistribution of poly(lactide-co-glycolide) nanospheres surface modified by poloxamer and poloxamine copolymers. J. Control. Release 1997, 44, 65–76. [Google Scholar] [CrossRef]
- Tabata, Y.; Murakami, Y.; Ikada, Y. Tumor accumulation of poly(vinyl alcohol) of different sizes after intravenous injection. J. Control. Release 1998, 50, 123–133. [Google Scholar] [CrossRef]
- Mosqueira, V.C.; Legrand, P.; Gref, R.; Heurtault, B.; Appel, M.; Barratt, G. Interactions between a macrophage cell line (J774A1) and surface-modified poly (D,L-lactide) nanocapsules bearing poly(ethylene glycol). J. Drug Target. 1999, 7, 65–78. [Google Scholar] [CrossRef]
- Zambaux, M.F.; Bonneaux, F.; Gref, R.; Dellacherie, E.; Vigneron, C. MPEO-PLA nanoparticles: effect of MPEO content on some of their surface properties. J. Biomed. Mater. Res. 1999, 44, 109–115. [Google Scholar] [CrossRef]
- Nguyen, C.A.; Allemann, E.; Schwach, G.; Doelker, E.; Gurny, R. Cell interaction studies of PLA-MePEG nanoparticles. Int. J. Pharm. 2003, 254, 69–72. [Google Scholar] [CrossRef]
- Yoshioka, M.; Hashida, M.; Muranihsi, S.; Sezaki, H. Specific delivery of mitomycin C to the liver, spleen and lung: nano- and microspherical carriers of gelatin. Int. J. Pharm. 1981, 81, 131–141. [Google Scholar]
- Leopold, C.S.; Friend, D.R. In vivo pharmacokinetic study for the assessment of poly(L-aspartic acid) as a drug carrier for colon-specific drug delivery. J. Pharmacokinet. Biopharm. 1995, 23, 397–406. [Google Scholar] [CrossRef]
- Mehvar, R.; Dann, R.O.; Hoganson, D.A. Kinetics of hydrolysis of dextran-methylprednisolone succinate, a macromolecular prodrug of methylprednisolone, in rat blood and liver lysosomes. J. Control. Release 2000, 68, 53–61. [Google Scholar]
- Rensberger, K.L.; Hoganson, D.A.; Mehvar, R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: in vitro immunosuppressive effects on rat blood and spleen lymphocytes. Int. J. Pharm. 2000, 207, 71–76. [Google Scholar] [CrossRef]
- Zhang, X.; Mehvar, R. Dextran-methylprednisolone succinate as a prodrug of methylprednisolone: plasma and tissue disposition. J. Pharm. Sci. 2001, 90, 2078–2087. [Google Scholar] [CrossRef]
- Yano, H.; Hirayama, F.; Kamada, M.; Arima, H,; Uekama, K. Colon-specific delivery of prednisolone-appended alpha-cyclodextrin conjugate: alleviation of systemic side effect after oral administration. J. Control. Release 2002, 79, 103–112. [Google Scholar] [CrossRef]
- Tozaki, H.; Fujita, T.; Komoike, J.; Kim, S.I.; Terashima, H.; Muranishi, S.; Okabe, S.; Yamamoto, A. Colon-specific delivery of budesonide with azopolymer-coated pellets: therapeutic effects of budesonide with a novel dosage form against 2,4,6-trinitrobenzenesulphonic acid-induced colitis in rats. J. Pharm. Pharmacol. 1999, 51, 257–261. [Google Scholar] [CrossRef]
- Nakase, H.; Okazaki, K.; Tabata, Y.; Uose, S.; Ohana, M.; Uchida, K.; Matsushima, Y.; Kawanami, C.; Oshima, C.; Ikada, Y.; Chiba, T. Development of an oral drug delivery system targeting immune-regulating cells in experimental inflammatory bowel disease: a new therapeutic strategy. J. Pharmacol. Exp. Ther. 2000, 292, 15–21. [Google Scholar]
- Lamprecht, A.; Ubrich, N.; Yamamoto, H.; Schäfer, U.; Takeuchi, H.; Maincent, P.; Kawashima, Y.; Lehr, C.M. Biodegradable nanoparticles for targeted drug delivery in treatment of inflammatory bowel disease. J. Pharmacol. Exp. Ther. 2001, 299, 775–781. [Google Scholar]
- Tozaki, H.; Odoriba, T.; Okada, N.; Fujita, T.; Terabe, A.; Suzuki, T.; Okabe, S.; Muranishi, S.; Yamamoto, A. Chitosan capsules for colon-specific drug delivery: enhanced localization of 5-aminosalicylic acid in the large intestine accelerates healing of TNBS-induced colitis in rats. J. Control. Release 2002, 82, 51–61. [Google Scholar] [CrossRef]
- Onishi, H.; Oosegi, T.; Machida, Y.; McGinity, J.W. Preparation and in vitro evaluation of chitosan microspheres containing prednisolone: comparison of simple and conjugate microspheres. Drug Dev. Ind. Pharm. 2005, 31, 597–605. [Google Scholar] [CrossRef]
- Onishi, H.; Oosegi, T.; Machida, Y. Efficacy and toxicity of Eudragit-coated chitosan-succinyl-prednisolone conjugate microspheres using rats with 2, 4, 6-trinitrobenzenesulfonic acid-induced colitis. Int. J. Pharm. 2008, 358, 271–277. [Google Scholar] [CrossRef]
- Oosegi, T.; Onishi, H.; Machida, Y. Gastrointestinal distribution and absorption behavior of Eudragit-coated chitosan-prednisolone conjugate microspheres in rats with TNBS-induced colitis. Int. J. Pharm. 2008, 348, 80–88. [Google Scholar] [CrossRef]
- Sato, M.; Onishi, H.; Takahara, J.; Machida, Y.; Nagai, T. In vivo drug release and antitumor characteristics of water-soluble conjugates of mitomycin C with glycol-chitosan and N-succinyl-chitosan. Biol. Pharm. Bull. 1996, 19, 1170–1177. [Google Scholar] [CrossRef]
- Sato, M.; Onishi, H.; Kitano, M.; Machida, Y.; Nagai, T. Preparation and drug release characteristics of the conjugates of mitomycin C with glycol-chitosan and N-succinyl-chitosan. Biol. Pharm. Bull. 1996, 19, 241–245. [Google Scholar] [CrossRef]
- Song, Y.; Onishi, H.; Nagai, T. Synthesis and drug-release characteristics of the conjugates of mitomycin C with N-succinyl-chitosan and carboxymethyl-chitin. Chem. Pharm. Bull. 1992, 40, 2822–2825. [Google Scholar] [CrossRef]
- Song, Y.; Onishi, H.; Nagai, T. Pharmacokinetic characteristics and antitumor activity of the N-succinyl-chitosan-mitomycin C conjugate and the carboxymethyl-chitin-mitomycin C conjugate. Biol. Pharm. Bull. 1993, 16, 48–54. [Google Scholar] [CrossRef]
- Song, Y.; Onishi, H; Machida, Y.; Nagai, T. Particle characteristics of carboxymethyl-chitin-mitomycin C conjugate and N-succinyl-chitosan-mitomycin C conjugate and their distribution and histological effect in some tissues. S. T. P. Pharma Sci. 1995, 5, 162–170. [Google Scholar]
- Onishi, H.; Takahashi, H.; Yoshiyasu, M.; Machida, Y. Preparation and in vitro properties of N-succinylchitosan- or carboxymethylchitin-mitomycin C conjugate microparticles with specified size. Drug Dev. Ind. Pharm. 2001, 27, 659–667. [Google Scholar] [CrossRef]
- Onishi, H.; Kume, K.; Koyama, K.; Machida, Y. Preparation of carboxymethylchitin nanoparticles by covalent crosslinking and their in vitro evaluation. Open Drug Deliv. J. 2008, 2, 20–25. [Google Scholar] [CrossRef]
- Okada, H.; Heya, T.; Ogawa, Y.; Shimamoto, T. One-month release injectable microcapsules of a luteinizing hormone-releasing hormone agonist (leuprolide acetate) for treating experimental endometriosis in rats. J. Pharmacol. Exp. Ther. 1988, 244, 744–750. [Google Scholar]
- Ogawa, Y.; Okada, H.; Yamamoto, M.; Shimamoto, T. In vivo release profiles of leuprolide acetate from microcapsules prepared with polylactic acids or copoly(lactic/glycolic) acids and in vivo degradation of these polymers. Chem. Pharm. Bull. 1988, 36, 2576–2781. [Google Scholar] [CrossRef]
- Machida, Y.; Onishi, H.; Kurita, A.; Hata, H.; Morikawa, A.; Machida, Y. Pharmacokinetics of prolonged-release CPT-11-loaded microspheres in rats. J. Control. Release 2000, 66, 159–175. [Google Scholar] [CrossRef]
- Takahashi, H.; Onishi, H.; Machida, Y. Glycyrrhetic acid-loaded microparticles: liver-specific delivery and therapeutic potential against carbon tetrachloride-induced hepatitis. J. Pharm. Pharmacol. 2004, 56, 437–444. [Google Scholar] [CrossRef]
- Oh, J.E; Nam, Y.S.; Lee, K.H.; Park, T.G. Conjugation of drug to poly(D,L-lactic-co-glycolic acid) for controlled release from biodegradable microspheres. J. Control. Release 1999, 57, 269–280. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly(-lactic acid) microspheres: effect of molecular weight. J. Control. Release 1994, 30, 161–173. [Google Scholar]
- Yoo, H.S.; Oh, J.E.; Lee, K.H.; Park, T.G. Biodegradable nanoparticles containing doxorubicin-PLGA conjugate for sustained release. Pharm. Res. 1999, 16, 1114–1118. [Google Scholar] [CrossRef]
- Yoo, H.S.; Lee, K.H.; Oh, J.E.; Park, T.G. In vitro and in vivo anti-tumor activities of nanoparticles based on doxorubicin-PLGA conjugates. J. Control. Release 2000, 68, 419–431. [Google Scholar] [CrossRef]
- Fiocchi, C. Inflammatory bowel disease. Current concepts of pathogenesis and implications for therapy. Minerva Gastroenterol. Dietol. 2002, 48, 215–226. [Google Scholar]
- Campieri, M.; Ferguson, A.; Doe, W.; Persson, T.; Nilsson, L.G. Oral budesonide is as effective as oral prednisolone in active Crohn's disease. The Global Budesonide Study Group.. Gut 1997, 41, 209–214. [Google Scholar] [CrossRef]
- Rutgeerts, P. The use of oral topically acting glucocorticosteroids in the treatment of inflammatory bowel disease. Mediators Inflamm. 1998, 7, 137–140. [Google Scholar] [CrossRef]
- Friend, D.R. New oral delivery systems for treatment of inflammatory bowel disease. Adv. Drug Deliv. Rev. 2005, 57, 247–265. [Google Scholar] [CrossRef]
- Lamprecht, A.; Schafer, U.; Lehr, C.M. Microparticle targeting to the inflamed colonic mucosa for the treatment of inflammatory bowel disease: in vivo results from rat. AAPS PharmSci. 2000, 2(S1), 1930. [Google Scholar]
- Lamprecht, A.; Schafer, U.; Lehr, C.M. Size-dependent bioadhesion of micro- and nanoparticulate carriers to the inflamed colonic mucosa. Pharm. Res. 2001, 18, 788–793. [Google Scholar] [CrossRef]
- Tabata, Y.; Ikada, Y. Effect of the size and surface charge of polymer microspheres on their phagocytosis by macrophage. Biomaterials 1988, 9, 356–362. [Google Scholar] [CrossRef]
- Tabata, Y.; Inoue, Y.; Ikada, Y. Size effect on systemic and mucosal immune responses induced by oral administration of biodegradable microspheres. Vaccine 1996, 14, 1677–1685. [Google Scholar] [CrossRef]
- Oosegi, T.; Onishi, H.; Machida, Y. Novel preparation of enteric-coated chitosan-prednisolone conjugate microspheres and in vitro evaluation of their potential as a colonic delivery system. Eur. J. Pharm. Biopharm. 2008, 68, 260–266. [Google Scholar] [CrossRef]
- Silva, M.; Lara, A.S.; Leite, C.Q.; Ferreira, E.I. Potential tuberculostatic agents: micelle-forming copolymer poly(ethylene glycol)-poly(aspartic acid) prodrug with isoniazid. Arch. Pharm. (Weinheim) 2001, 334, 189–193. [Google Scholar] [CrossRef]
- Hans, M.; Shimoni, K.; Danino, D.; Siegel, S.J.; Lowman, A. Synthesis and characterization of mPEG-PLA prodrug micelles. Biomacromolecules 2005, 6, 2708–2717. [Google Scholar] [CrossRef]
- Silva, M.; Ricelli, N.L.; El Seoud, O.; Valentim, C.S.; Ferreira, A.G.; Sato, D.N.; Leite, C.Q.F.; Ferreira, E.I. Potential tuberculostatic agent: micelle-forming pyrazinamide prodrug. Arch. Pharm. (Weinheim) 2006, 339, 283–290. [Google Scholar] [CrossRef]
- Silva, M.; Ferreira, E.I.; Leite, C.Q.F.; Sato, D.N. Preparation of polymeric micelles for use as carriers of tuberculostatic drugs. Trop. J. Pharm. Res. 2007, 6, 815–824. [Google Scholar]
- Li, X.; Wu, Q.; Lu, M.; Zhang, F.; Lin, X. Novel hepatoma-targeting micelles based on chemoenzymatic synthesis and self-assembly of galactose-functionalized ribavirin-containing amphophilic random copolymer. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 2734–2744. [Google Scholar] [CrossRef]
© 2008 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Onishi, H.; Machida, Y. In Vitro and In Vivo Evaluation of Microparticulate Drug Delivery Systems Composed of Macromolecular Prodrugs. Molecules 2008, 13, 2136-2155. https://doi.org/10.3390/molecules13092136
Onishi H, Machida Y. In Vitro and In Vivo Evaluation of Microparticulate Drug Delivery Systems Composed of Macromolecular Prodrugs. Molecules. 2008; 13(9):2136-2155. https://doi.org/10.3390/molecules13092136
Chicago/Turabian StyleOnishi, Hiraku, and Yoshiharu Machida. 2008. "In Vitro and In Vivo Evaluation of Microparticulate Drug Delivery Systems Composed of Macromolecular Prodrugs" Molecules 13, no. 9: 2136-2155. https://doi.org/10.3390/molecules13092136
APA StyleOnishi, H., & Machida, Y. (2008). In Vitro and In Vivo Evaluation of Microparticulate Drug Delivery Systems Composed of Macromolecular Prodrugs. Molecules, 13(9), 2136-2155. https://doi.org/10.3390/molecules13092136