“Flash” Solvent-free Synthesis of Triazoles Using a Supported Catalyst

Abstract
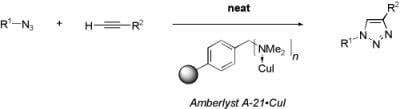
:
Introduction

Results and Discussion


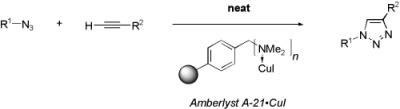{kind=link}
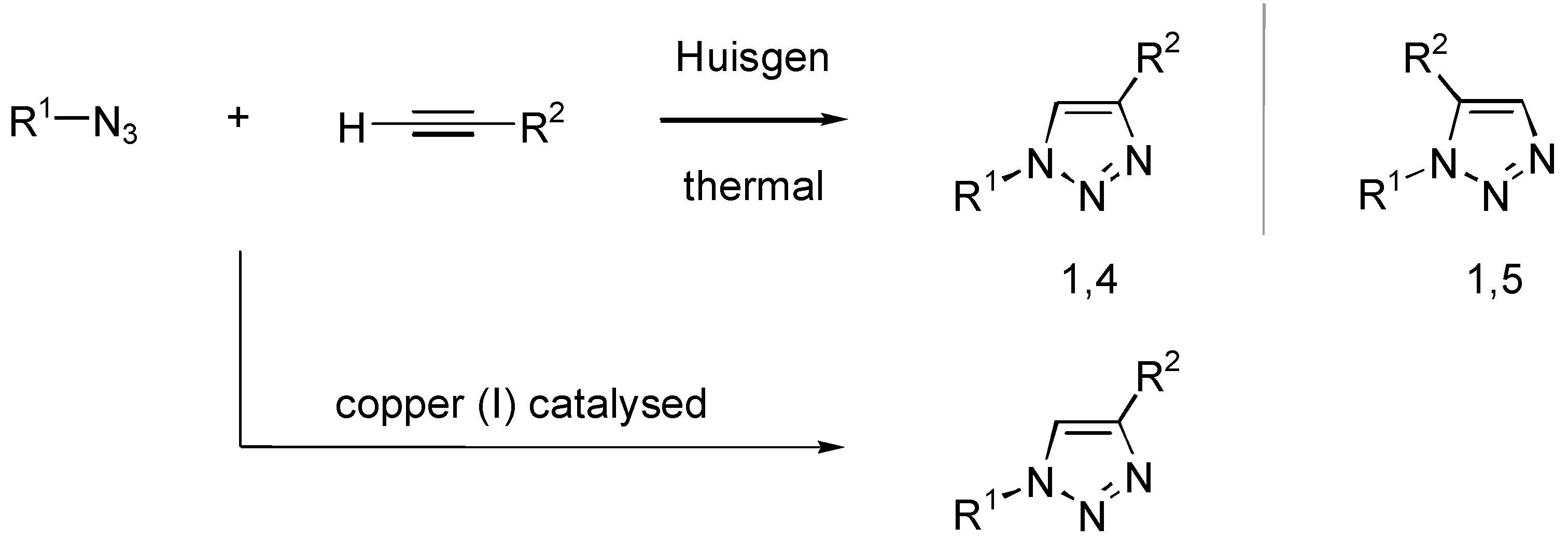{kind=link}
{kind=link}
 |  |  |  |  |  | |
|---|---|---|---|---|---|---|
| 2a | 2b | 2c | 2d[b] | 2e | 2f[c] | |
| PhCH2–N3 | 99 | 95 | 99 | 99 | 99 | 99 |
| 1a | ||||||
 | 93 | 68 | 76 | 72 | 99 | 72 |
| 1b | ||||||
 | 99 | 92 | 99 | 96 | 99 | 83 |
| 1c | ||||||
 | 76 | 89 | 98 | 90 | 86 | 90 |
| 1d |
Conclusions
Experimental
General
Procedures
General procedure for triazole synthesis
Acknowledgements
References and Notes
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar Cycloadditions. Past and Future. [CrossRef]
- Tornøe, C.W.; Meldal, M. Peptidotriazoles: Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions on Solid phase. In 17th American Peptides Symposium Proceedings Book. Peptides: The Wave of the Future; Lebl, M., Houghten, R.A., Eds.; American Peptide Society and Kluwer Academic: San Diego, USA, 2001; pp. 263–264. [Google Scholar]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes.
- Bock, V. D.; Hiemstra, H.; van Maarseveen, J. H. CuI-Catalyzed Alkyne-Azide Click Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 51–68, and references [9,10]. [Google Scholar]
- Wu, P.; Fokin, V.V. Catalytic Azide–Alkyne Cycloaddition: Reactivity and Applications. Aldrichim. Acta 2007, 40, 7–17. [Google Scholar]
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide−Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef]
- Zhang, X.; Hsung, R.P.; Li, H. A triazole-templated ring-closing metathesis for constructing novel fused and bridged triazoles. Chem. Commun. 2007, 2420–2422, and references [12,13]. [Google Scholar] [CrossRef]
- Bertrand, P.; Gesson, J.-P. Click Chemistry with O-Dimethylpropargylcarbamate for Preparation of pH-Sensitive Functional Groups. A Case Study. J. Org. Chem. 2007, 72, 3596–3599. [Google Scholar] [CrossRef]
- Peddibhotla, S.; Dang, Y.; Liu, J.O.; Romo, D. Simultaneous Arming and Structure/Activity Studies of Natural Products Employing O−H Insertions: An Expedient and Versatile Strategy for Natural Products-Based Chemical Genetics. J. Am. Chem. Soc. 2007, 129, 12222–12231. [Google Scholar] [CrossRef]
- Beckmann, H.S.G.; Wittmann, V. One-Pot Procedure for Diazo Transfer and Azide−Alkyne Cycloaddition: Triazole Linkages from Amines. Org. Lett. 2007, 9, 1–4, and references [15,16]. [Google Scholar] [CrossRef]
- Tao, C.-Z.; Cui, X.; Li, J.; Liu, A.-X.; Liu, L.; Guo, Q.-X. Copper-catalyzed synthesis of aryl azides and 1-aryl-1,2,3-triazoles from boronic acids. Tetrahedron Lett. 2007, 48, 3525–3529. [Google Scholar] [CrossRef]
- Barral, K.; Moorhouse, A.D.; Moses, J.E. Efficient Conversion of Aromatic Amines into Azides: A One-Pot Synthesis of Triazole Linkages. Org. Lett. 2007, 9, 1809–1811. [Google Scholar] [CrossRef]
- Chassaing, S.; Kumarraja, M.; Sani Souna Sido, A.; Pale, P.; Sommer, J. Click Chemistry in CuI-zeolites: The Huisgen [3 + 2]-Cycloaddition. Org. Lett. 2007, 9, 883–886. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Taft, B.R. Heterogeneous Copper-in-Charcoal-Catalyzed Click Chemistry. [CrossRef]
- Jlalia, I.; Elamari, H.; Meganem, F.; Herscovici, J.; Girard, C. Copper(I)-doped Wyoming’s montmorillonite for the synthesis of disubstituted 1,2,3-triazoles. Tetrahedron Lett. 2008, 49, 6756–6758. [Google Scholar] [CrossRef]
- Appukkuttan, P.; Dehaen, W.; Fokin, V.V.; Van der Eycken, E. A Microwave-Assisted Click Chemistry Synthesis of 1,4-Disubstituted 1,2,3-Triazoles via a Copper(I)-Catalyzed Three-Component Reaction. Org. Lett. 2004, 6, 4223–4225, and references [21,22] therein. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar]
- David, O.; Maisonneuve, S.; Xie, J. Generation of new fluorophore by Click chemistry: synthesis and properties of β-cyclodextrin substituted by 2-pyridyl triazole. Tetrahedron Lett. 2007, 48, 6527–6530. [Google Scholar] [CrossRef]
- Durán Pachón, L.; van Maarseveen, J.H.; Rothenberg, G. Click Chemistry: Copper Clusters Catalyse the Cycloaddition of Azides with Terminal Alkynes. Adv. Synth. Catal. 2005, 347, 811–815. [Google Scholar] [CrossRef]
- Molteni, G.; Bianchi, C.L.; Marinoni, G.; Santo, N.; Ponti, A. Cu/Cu-oxide nanoparticles as catalyst in the click azide–alkyne cycloaddition. New J. Chem. 2006, 30, 1137–1139. [Google Scholar]
- Girard, C.; Önen, E.; Aufort, M.; Beauvière, S.; Samson, E.; Herscovici, J. Reusable Polymer-Supported Catalyst for the [3+2] Huisgen Cycloaddition in Automation Protocols. Org. Lett. 2006, 8, 1689–1692. [Google Scholar] [CrossRef]
- Chan, T.R.; Fokin, V.V. Polymer-Supported Copper(I) Catalysts for the Experimentally Simplified Azide–Alkyne Cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Walsh, P.J.; Li, H.; Anaya de Parrodi, C. A Green Chemistry Approach to Asymmetric Catalysis: Solvent-Free and Highly Concentrated Reactions. Chem. Rev. 2007, 107, 2503–2545. [Google Scholar] [CrossRef]
- Díez-González, S.; Correa, A.; Cavallo, L.; Nolan, S.P. (NHC)Copper(I)-Catalyzed [3 + 2] Cycloaddition of Azides and Mono- or Disubstituted Alkynes. Chem. Eur. J. 2006, 12, 7558–7564, and references [29,30,31] therein. [Google Scholar] [CrossRef]
- Guezguez, R.; Bougrin, K.; El Akri, K.; Benhida, R. A highly efficient microwave-assisted solvent-free synthesis of α- and β-2′-deoxy-1,2,3-triazolyl-nucleosides. Tetrahedron Lett. 2006, 47, 4807–4811. [Google Scholar] [CrossRef]
- El Akri, K.; Bougrin, K.; Balzarini, J.; Faraj, A.; Benhida, R. Efficient synthesis and in vitro cytostatic activity of 4-substituted triazolyl-nucleosides. Bioorg. Med. Chem. Lett. 2007, 17, 6656–6659. [Google Scholar] [CrossRef]
- Li, P.; Wang, L.; Zhang, Y. SiO2–NHC–Cu(I): an efficient and reusable catalyst for [3+2] cycloaddition of organic azides and terminal alkynes under solvent-free reaction conditions at room temperature. Tetrahedron 2008, 64, 10825–10830. [Google Scholar] [CrossRef]
- Smith, C.D.; Baxendale, I.R.; Lanners, S.; Hayward, J.J.; Smith, S.C.; Ley, S.V. [3 + 2] Cycloaddition of acetylenes with azides to give 1,4-disubstituted 1,2,3-triazoles in a modular flow reactor. Org. Biomol. Chem. 2007, 5, 1559–1561. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Singh, S.K. Synthesis of C-Carbamoyl-1,2,3-triazoles by Microwave-Induced 1,3-Dipolar Cycloaddition of Organic Azides to Acetylenic Amides. J. Org. Chem. 2002, 67, 9077–9079. [Google Scholar] [CrossRef]
- Pourcelot, G.; Cadiot, P. Preparation of propargylic, allenic and acetylenic derivatives of the elements of Group VIB. Bull. Soc. Chim. Fr. 1966, 3016–3024. [Google Scholar]
- Alvarez, S.G.; Alvarez, M.T. A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield. Synthesis 1997, 413–414. [Google Scholar] [CrossRef]
- Hooper, N.; Beeching, L.J.; Dyke, J.M.; Morris, A.; Ogden, J.S.; Dias, A.A.; Costa, M.L.; Barros, M.T.; Cabrell, M.H.; Moutinho, A.M.C. A Study of the Thermal Decomposition of 2-Azidoethanol and 2-Azidoethylacetate by Ultraviolet PES and Matrix Isolation Spectroscopy. J. Phys. Chem. A. 2002, 106, 9968–9975. [Google Scholar] [CrossRef]
- Scheel, A.J.; Komber, H.; Voit, B.I. Novel Hyperbranched Poly([1,2,3]-triazole)s Derived from AB2 Monomers by a 1,3-Dipolar Cycloaddition. Macromol. Rapid Commun. 2004, 25, 1175–1180. [Google Scholar] [CrossRef]
- Carboni, B.; Benalil, A.; Vaultier, M. Aliphatic amino azides as key building blocks for efficient polyamine syntheses. J. Org. Chem. 1993, 58, 3736–3741. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jlalia, I.; Meganem, F.; Herscovici, J.; Girard, C. “Flash” Solvent-free Synthesis of Triazoles Using a Supported Catalyst. Molecules 2009, 14, 528-539. https://doi.org/10.3390/molecules14010528
Jlalia I, Meganem F, Herscovici J, Girard C. “Flash” Solvent-free Synthesis of Triazoles Using a Supported Catalyst. Molecules. 2009; 14(1):528-539. https://doi.org/10.3390/molecules14010528
Chicago/Turabian StyleJlalia, Ibtissem, Faouzi Meganem, Jean Herscovici, and Christian Girard. 2009. "“Flash” Solvent-free Synthesis of Triazoles Using a Supported Catalyst" Molecules 14, no. 1: 528-539. https://doi.org/10.3390/molecules14010528
APA StyleJlalia, I., Meganem, F., Herscovici, J., & Girard, C. (2009). “Flash” Solvent-free Synthesis of Triazoles Using a Supported Catalyst. Molecules, 14(1), 528-539. https://doi.org/10.3390/molecules14010528