1. Introduction
Aptamers are oligonucleotide or peptide molecules that have the ability to bind to specific target molecules. Aptamers are usually created by selecting them from a huge random sequence pool, although some “natural” aptamers also exist (e.g. riboswitches). These molecules can be used for both basic research and clinical purposes as drugs and diagnostic sensors. Oligonucleotide aptamers can be identified through repeated rounds of
in vitro selection (SELEX, Systematic Evolution of Ligands by Exponential Enrichment), for binding to various molecular targets such as small molecules, proteins, nucleic acids, and even cells, tissues and organisms. Peptide aptamers are combinatorial protein reagents that are usually selected from randomized expression libraries in yeast, by virtue of their ability to bind to a given target protein under intracellular conditions. Most commonly, the term aptamer refers to oligonucleotide (RNA or DNA) aptamers, which is how we use the term here. Aptamers offer molecular recognition properties that rival those of antibodies. In addition to their high specificity and binding affinity [
1], they also offer many advantages over antibodies; they can be identified completely in vitro, are readily produced by chemical synthesis, possess desirable storage properties, are more robust for many processing conditions and thereby offer easy multiplexing capabilities, are much smaller than antibodies, and they elicit little or no immunogenicity in therapeutic applications.
In 1990, two labs independently developed the technique of selection [
2,
3]. The Gold lab coined the term SELEX for their process of selecting RNA ligands against T4 DNA polymerase. The Szostak lab, selecting RNA ligands against various organic dyes, coined the term aptamer (from the Latin word ‘aptus’ meaning to fit and Greek word ‘meros’ meaning particle) and described the process as
in vitro selection. Two years later, the Szostak lab and Gilead Sciences, independent of one another, used
in vitro selection schemes to evolve single stranded DNA ligands for organic dyes and the human coagulant thrombin, respectively [
4,
5]. There does not appear to be any systematic differences between RNA and DNA aptamers, although DNA aptamers have greater intrinsic chemical stability. In 2001, the SELEX process was automated by the Ellington lab and SomaLogic Inc, reducing the duration of a selection experiment from a few months to a few days [
6,
7]. Lately, the concept of smart aptamers has been introduced, which describes aptamers that are selected with pre-defined equilibrium (K
d) and rate (k
off, k
on) constants and thermodynamic (ΔH, ΔS) parameters of aptamer-target interaction [
8]. Kinetic capillary electrophoresis is the technology used for the selection of smart aptamers, and it yields aptamers after a few rounds of selection [
9].
While the process of artificially engineering nucleic acid ligands is very useful in biology and biotechnology, the notion of aptamers in the natural world was note appreciated until 2002, when the Breaker lab discovered a nucleic acid-based genetic regulatory element called a “riboswitch” that possesses similar molecular recognition properties to artificially made aptamers [
10,
11]. In addition to the discovery of a new mode of genetic regulation, this added further credence to the notion of an ‘RNA World’, a postulated stage of early life on earth [
12]. Recent developments in aptamer-based therapeutics have been rewarded in the form of the first aptamer-based drug approved by the US FDA for treatment of age-related macular degeneration (AMD), called Macugen offered by OSI Pharmaceuticals (
http://www.osip.com). Some other companies are very active within this field, having aptamer based therapeutics and/or diagnostics under product development or in clinical trials. Examples include Archemix Inc., USA (
http://www.archemix.com), Noxxon Pharma AG, Germany (
http://www.noxxon.net), Isis Innovation Ltd., UK (
http://www.isis-innovation.com), and SomaLogic Inc., USA (
http://www.somalogic.com). In 2008, a technology for biomarker discovery termed AptaBiD (Aptamer-Facilitated Biomarker Discovery) was described for definition of molecular targets on cells, which facilitates exponential detection of biomarkers [
13]. It involves three major stages: (i) differential multi-round selection of aptamers recognizing biomarkers on target cells; (ii) aptamer-based isolation of biomarkers from target cells; and (iii) mass spectrometric identification of biomarkers. The important feature of the AptaBiD technology is that it produces synthetic affinity probes (aptamers) simultaneously with biomarker discovery. In AptaBiD, aptamers are developed for cell surface biomarkers in their native state and conformation. In addition to facilitating biomarker identification, such aptamers can be directly used for cell isolation, cell visualization, and tracking cells in vivo. They can also be used to modulate activities of cell receptors and facilitate delivery of different reagents (e.g. siRNA and drugs) into the cells.
Within less than two decades, a large number of aptamer-related articles have been published (searching PubMed with the term “aptamers” yields > 1700 hits, and “DNA aptamers yields > 1200; see
www.ncbi.nlm.nih.gov), and many aspects of aptamer research and development have been thoroughly reviewed, including (i) SELEX technology, which includes the general principle, starting random DNA oligonucleotide libraries, targets, selection conditions, amplification, cloning and characterization; (ii) chemical modifications; (iii) fields of application; and (iv) advantages and limitations. This review is intended to get around a central limitation of the selection process, namely ways to reduce the influence of the constant primer regions of the nucleotide libraries that they are used for identification of aptamers. While elegant computational methods have suggested that fixed primer sequences do not significantly influence the secondary structure of already-identified aptamers [
14], the presence of fixed primer sequences during selection can result in many problems [
15,
16].
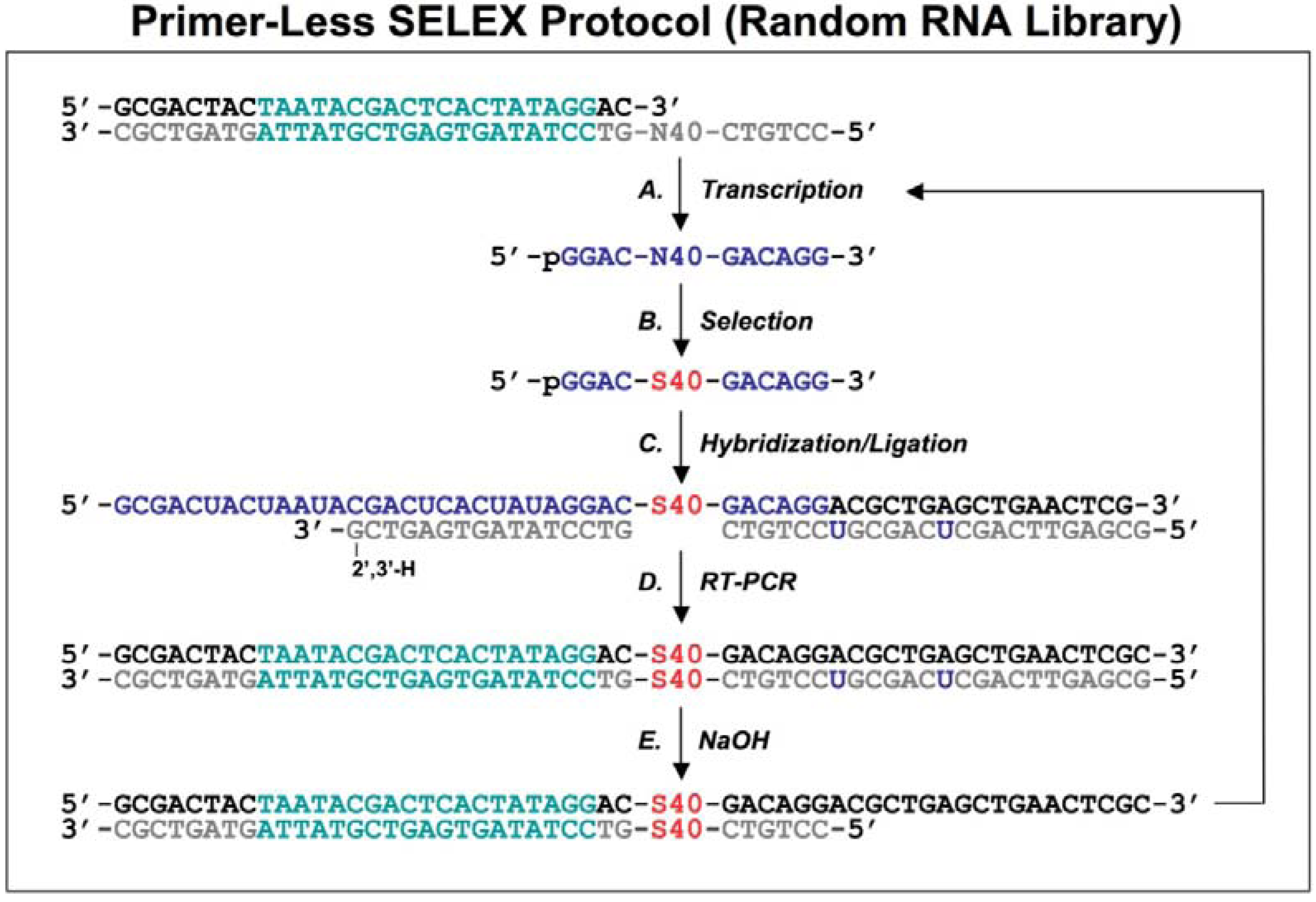
2. Primer-eliminating methods for genomic SELEX
In SELEX as originally developed [
2], the library utilized contained 10
14-10
15 random sequences. PCR amplification requires that the nucleic acid sequences of interest to be flanked by fixed sequence primer annealing sites. A T7 promoter is included in one of the primer annealing sides so that the library can be expressed as RNA, or a biotin is linked to a primer to allow use of a single stranded DNA library. Genomic SELEX is an extension of SELEX, and provides a useful tool for identifying genomic DNA or RNA transcripts that bind tightly to target proteins
in vitro, with the expectation that identified protein-nucleic acid interactions will provide insight into
in vivo biological events. To yield significant results, the binding between target proteins and the genomic library should not be influenced by artificial factors, such as the presence of primer-annealing sequences of the library. One important drawback is that the diversity of the genomic libraries is limited; for example, a fully representative genomic library of
Escherichia coli contains only ~4.6 ´ 10
6 different genomic sequences, although it is well-suited for the identification of specific nucleic acid binding protein recognition sites.
2.1. Primer-annealing and primer-switching genomic RNA library selection
Shtatland
et al. [
16] first reported 2 methods (termed “primer-annealing” and “primer-switching”) using an
E. coli genomic library constructed previously [
17].
E.coli genomic DNA was denatured and annealed to an oligo with 9 random nucleotides (termed ‘tail’) at the 3′ end and a fixed sequence at the 5′ end, then the oligo was extended with Klenow fragment of
E.coli DNA polymerase (
Figure 1A
1). Another randomized oligo with a different 5′ fixed sequence was added to the first reaction products, then annealed and extended in the same way (
Figure 1A2).
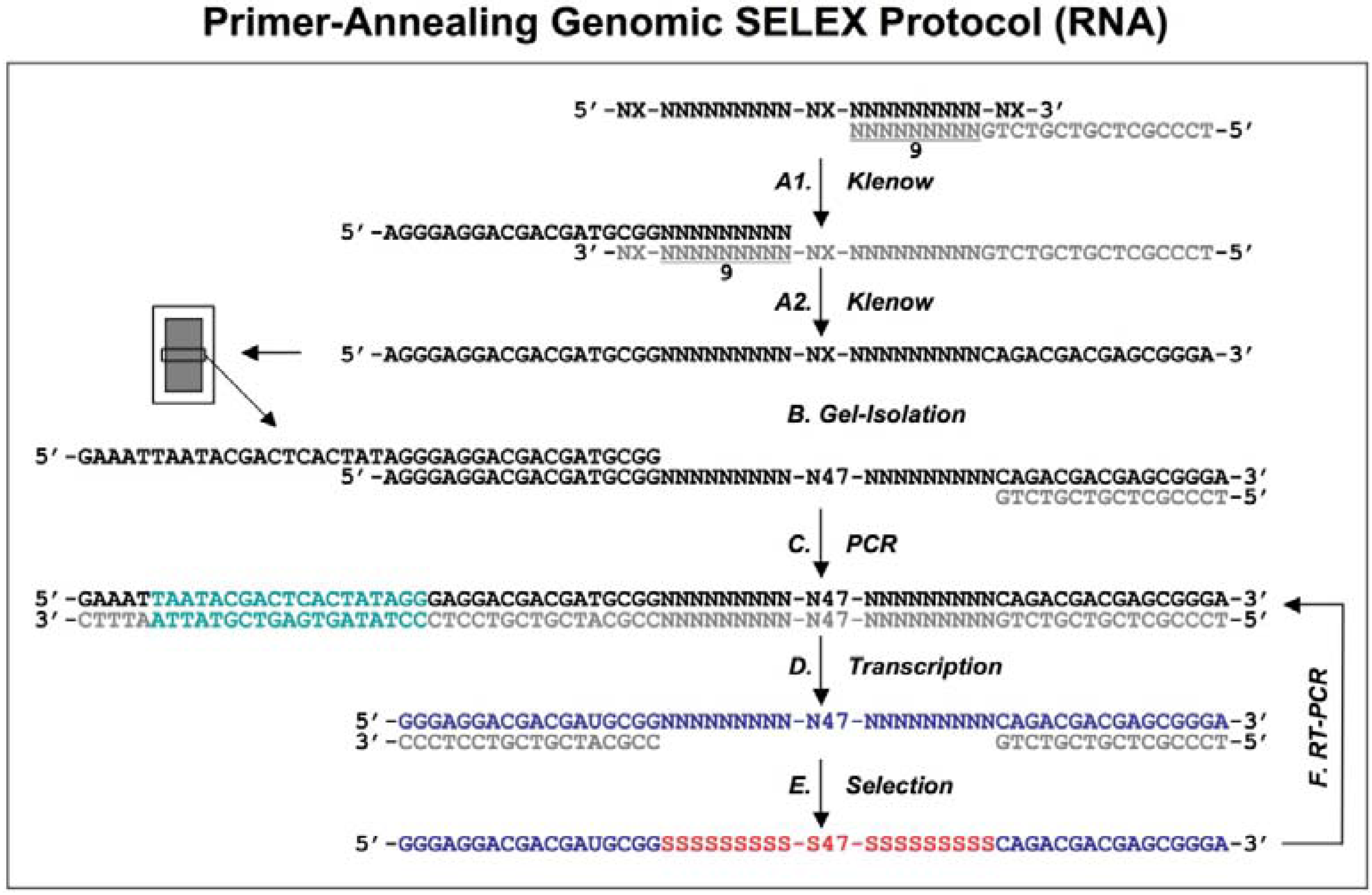
Figure 1.
Diagram of genomic library construction and primer-annealing genomic SELEX. The 3’ primer (indicated in gray) is annealed to the denatured genomic DNA (black), and the newly synthesized strand by Klenow enzyme (A1) serves as template for second strand synthesis (A2) primed by the 5’ primer (black). The products of the reactions are run on a denaturing gel and a fraction containing ~65 nt genomic inserts is isolated (B) and then PCR amplified to generate the
in vitro transcription templates (C; the green portion indicates a T7 RNA polymerase promoter). RNA library (blue) is transcribed (D), and subjected to the regular selection (without the complementary oligos, indicated in gray) and the primer-annealing selection (with the gray oligos) in presence of target protein (E). The target bound sequences (red) are amplified by RT-PCR for next round selection (F). This protocol is distilled from Shtatland
et al. [
16]
Figure 1.
Diagram of genomic library construction and primer-annealing genomic SELEX. The 3’ primer (indicated in gray) is annealed to the denatured genomic DNA (black), and the newly synthesized strand by Klenow enzyme (A1) serves as template for second strand synthesis (A2) primed by the 5’ primer (black). The products of the reactions are run on a denaturing gel and a fraction containing ~65 nt genomic inserts is isolated (B) and then PCR amplified to generate the
in vitro transcription templates (C; the green portion indicates a T7 RNA polymerase promoter). RNA library (blue) is transcribed (D), and subjected to the regular selection (without the complementary oligos, indicated in gray) and the primer-annealing selection (with the gray oligos) in presence of target protein (E). The target bound sequences (red) are amplified by RT-PCR for next round selection (F). This protocol is distilled from Shtatland
et al. [
16]
The extended reaction products were run on a denaturing gel to fractionate by size, and each fraction became the basis of a library with a different length of genomic insert (
Figure 1B). The library was completed by PCR amplification that added a T7 transcription promoter to one of the primer annealing sites (
Figure 1C). Finally, a library containing ~65 nt genomic inserts (9 nt tail + 47 nt genomic insert + 9 nt tail) flanked by fixed sequences, which served as primer annealing sites for amplification, was chosen for further primer-annealing SELEX (
Figure 1D-F) and primer-switching SELEX (
Figure 2). In each round of selection, the transcribed libraries were allowed to bind target (MS2 coat protein) and then the bound RNAs were amplified by RT-RCR.
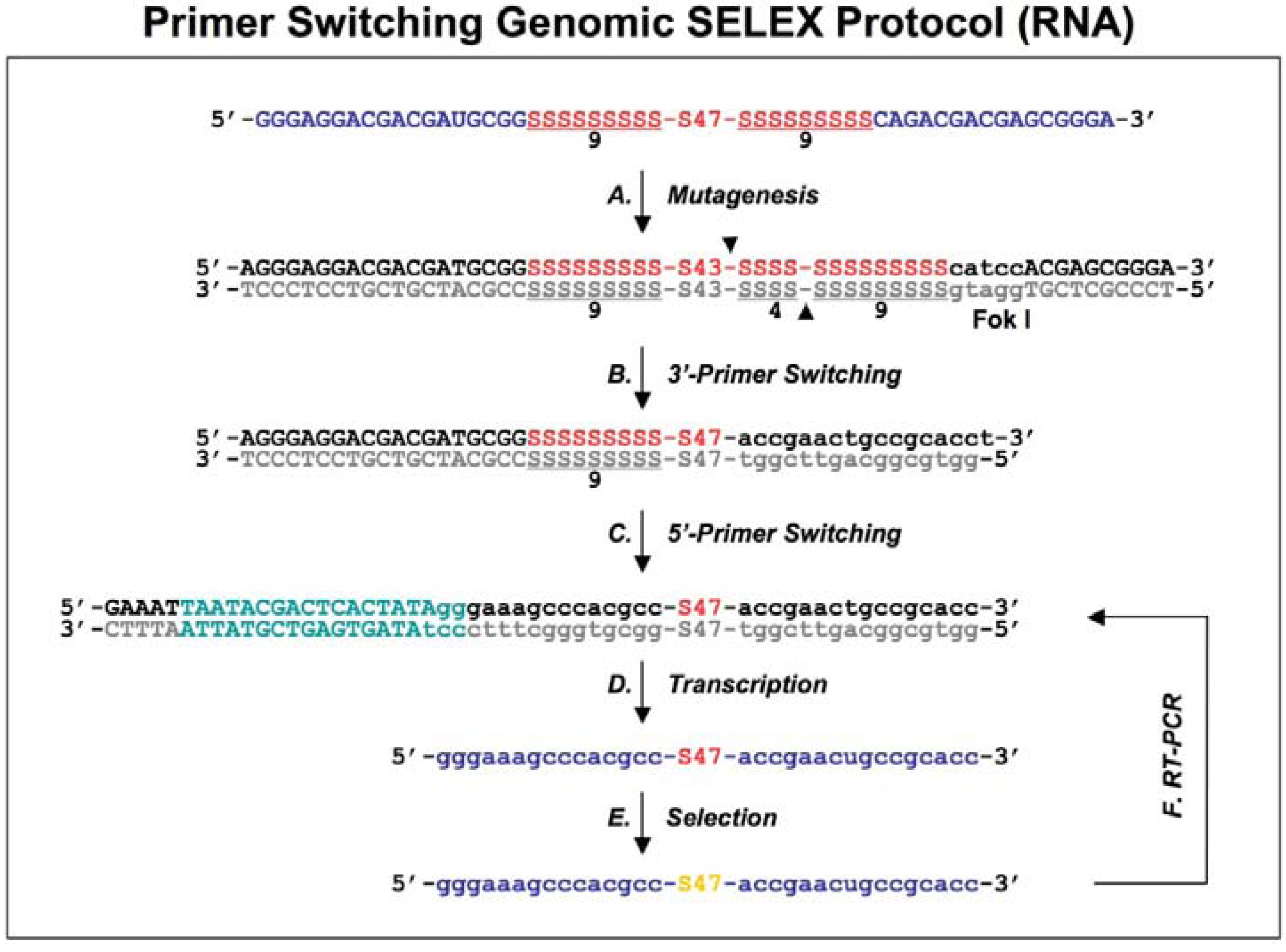
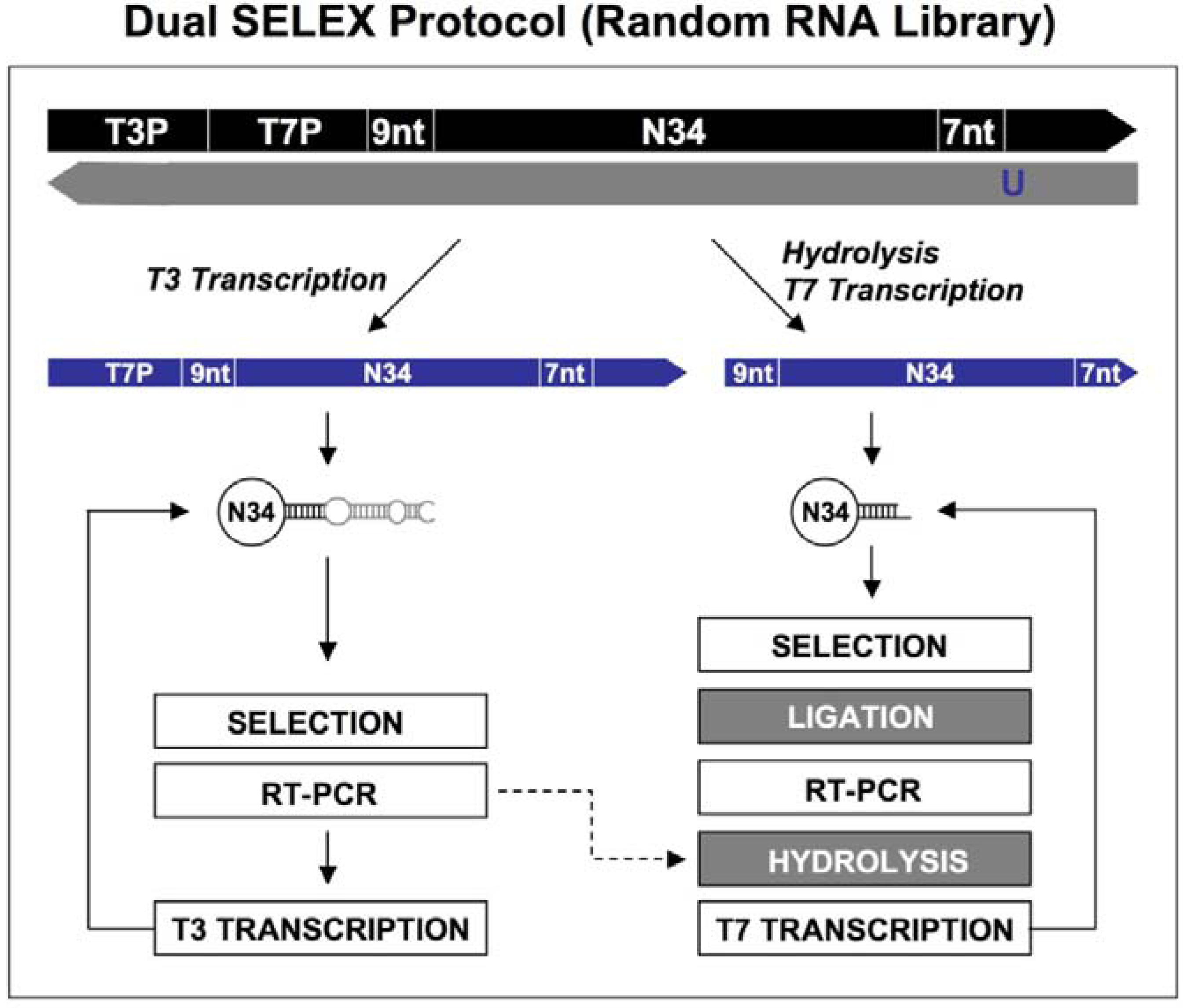
Figure 2.
Diagram of primer-switching genomic SELEX. The third round RNAs selected by regular selection are reverse transcribed to cDNAs, and PCR amplified by 5’primer and modified 3’ primer to a introduce Fok I restriction site (indicated in lower case, with its cutting sites indicated by black arrowheads; A). After Fok I digestion the overhang at the cut end of the library DNA is extended with Klenow enzyme, and blunt-end ligated to the new 3’ fixed sequence indicated in lower case (B). The steps of A and B are repeated but this time for the 5’ end, to generate a new transcription template (C; the T7 RNA polymerase promoter is indicated in green) for the following transcriptions, selections and re-amplifications (D-F). This protocol is distilled from Shtatland
et al . [
16]
Figure 2.
Diagram of primer-switching genomic SELEX. The third round RNAs selected by regular selection are reverse transcribed to cDNAs, and PCR amplified by 5’primer and modified 3’ primer to a introduce Fok I restriction site (indicated in lower case, with its cutting sites indicated by black arrowheads; A). After Fok I digestion the overhang at the cut end of the library DNA is extended with Klenow enzyme, and blunt-end ligated to the new 3’ fixed sequence indicated in lower case (B). The steps of A and B are repeated but this time for the 5’ end, to generate a new transcription template (C; the T7 RNA polymerase promoter is indicated in green) for the following transcriptions, selections and re-amplifications (D-F). This protocol is distilled from Shtatland
et al . [
16]
First, a standard SELEX protocol was performed (which was same as the primer-annealing SELEX shown in Steps D-F in
Figure 1, except that the complementary oligos of the fixed regions were absent during the selection process) with five rounds of selection. The second, primer-annealing SELEX, was designed to reduce the participation in binding of only the fixed sequences, but not the tails. The standard SELEX protocol was carried out for three rounds. In the three subsequent rounds, two DNA oligonucleotides complementary to the two fixed sequences were annealed to RNA prior to its binding to target protein. Switching from “no annealing” to “annealing”, rather than doing all six rounds “annealing”, should reduce the fraction of isolates that require annealing for binding to target protein. The third method, primer-switching SELEX (
Figure 2), eliminated the binding artifacts by completely switching the tails and fixed sequences halfway through the course of SELEX (replacing the fixed sequences with entirely different fixed ones). A FokI endonuclease restriction site (FokI cuts at a specific distance, 9-13 nt, from its restriction site) was introduced by RT-PCR (
Figure 2A) and used for cutting the 3′ fixed sequence and the tail of the library DNA selected after three rounds of regular SELEX. After digestion with FokI, the overhang at the cut end of the library DNA was extended with the Klenow fragment of
E.coli DNA polymerase, and blunt-end ligated to the new 3′ fixed sequence, which was a duplex of synthetic oligonucleotides (
Figure 2B). The ligation product was amplified by PCR and the whole procedure was repeated, this time to switch the 5′ fixed sequence (
Figure 2C).
The three pools of selected DNAs obtained from the three different protocols were cloned and sequenced after five or six rounds of selection, at which point optimal binding was observed. The results showed: (i) for the regular SELEX, 90% of the GeneBank related isolates were experimentally-induced artifacts; in most of them the fixed sequence participated in forming the binding site, which also had several mutations in the tails; (ii) for the primer-annealing SELEX, 60% of GenBank related isolates were artifacts, and the fraction of artifacts in which the fixed sequences participated in binding decreased ~2-fold; and (iii) for the primer-switching SELEX, only 1 out of 77 GeneBank related isolates was artifactual. These quantitative data clearly demonstrate the potential problems which can be caused by fixed-primer sequences.
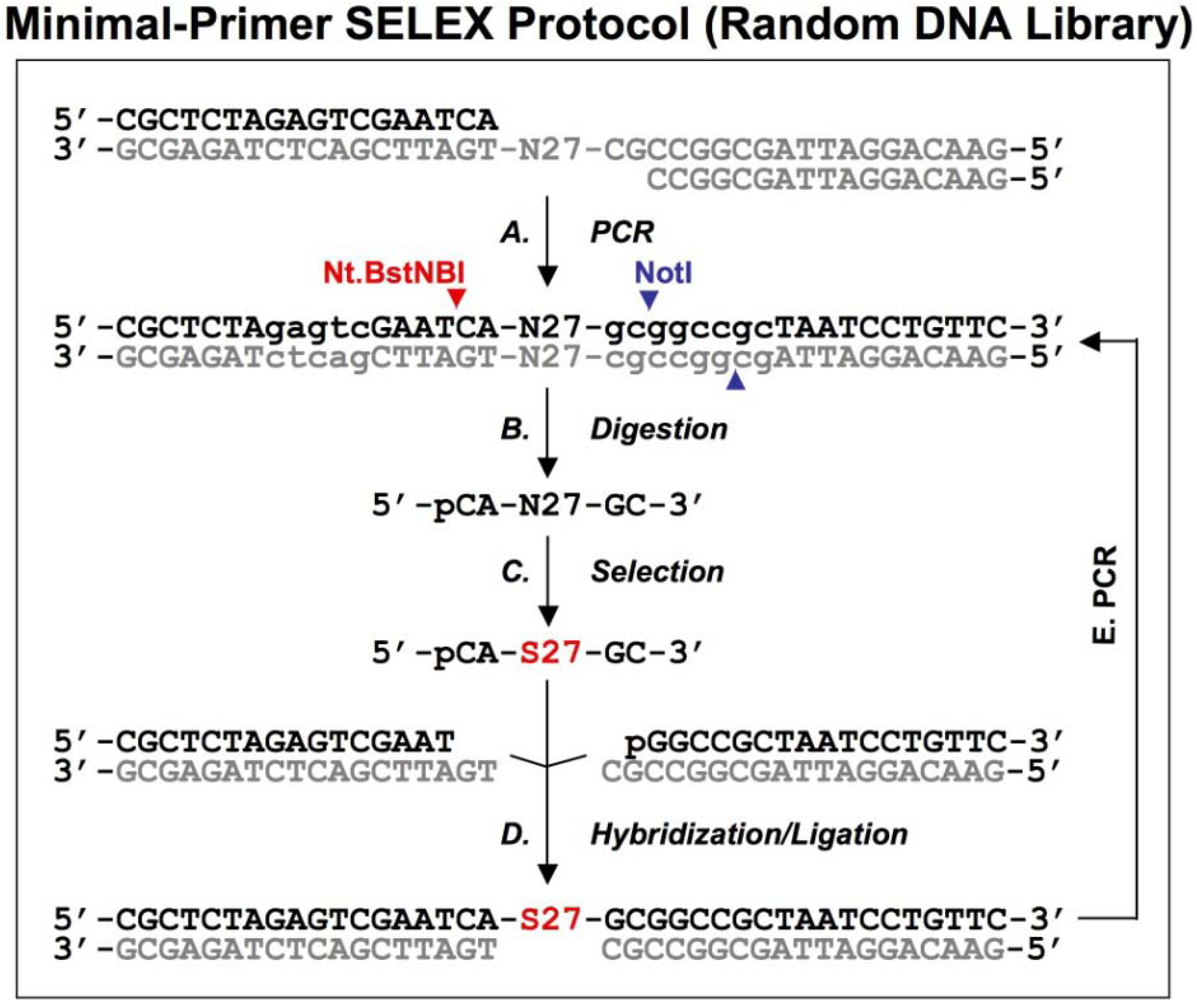
2.2. Primer-free genomic DNA library selection
To completely eliminate artifacts introduced by the fixed regions (including the tails and the primers), Wen and Gray designed a primer-free genomic SELEX method [
18], in which the primer-annealing sites were removed prior to the selection step and then regenerated prior to the amplification.
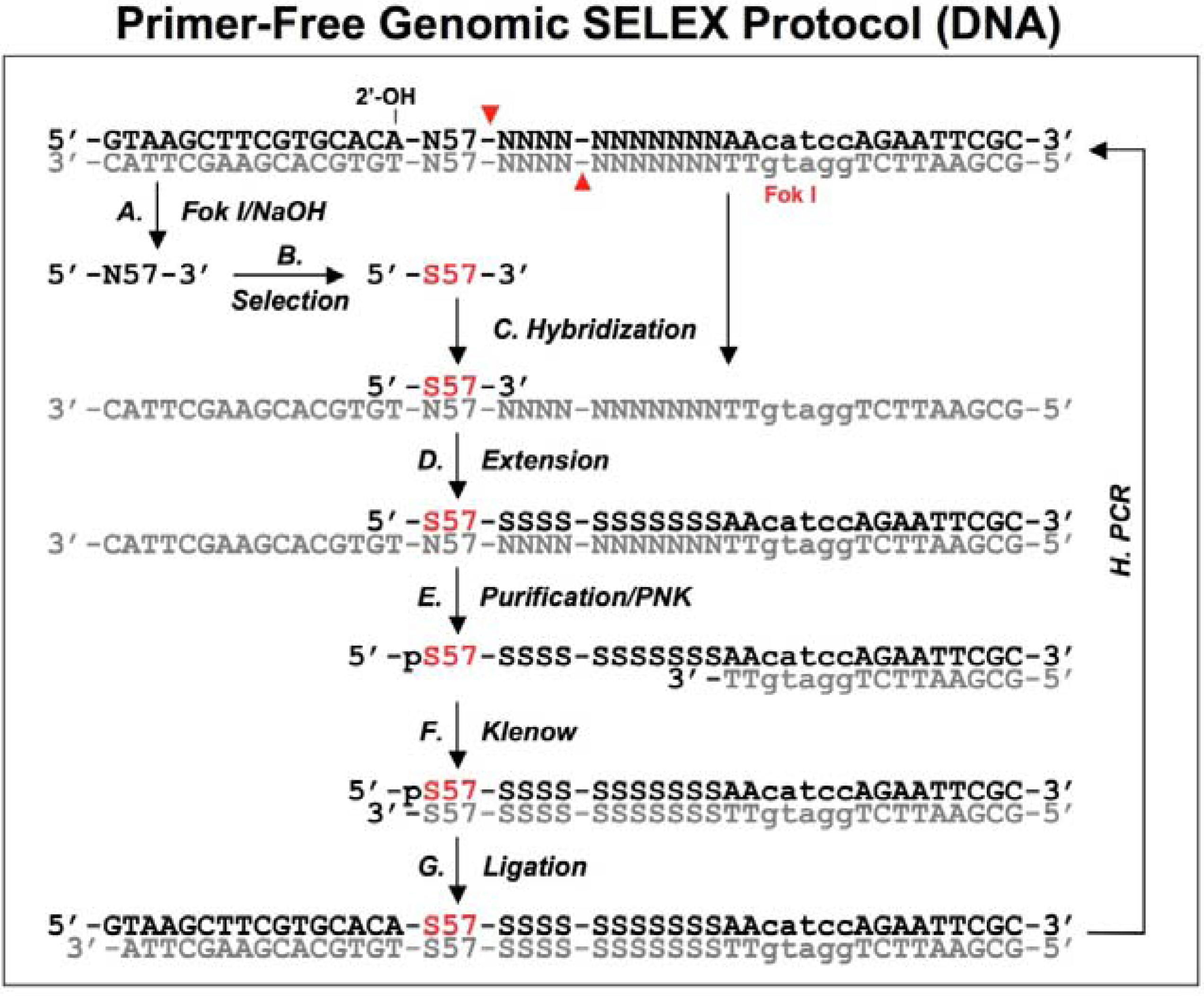
Figure 3.
A schematic procedure for primer-free genomic SELEX. The genomic library is shown as dsDNA, at the conjunction between 5’ primer and genomic insert a ribose is indicated as 2’-OH, at the 3’ end the Fok I recognition site is indicated as lower case and its cutting sites are indicated by the red arrowheads. The starting library is generated by Fok I digestion followed by alkaline hydrolysis (A), after selection (B), it is annealed to dsDNA library (C) to synthesize the 3’ tail and fixed sequences (D). The extended products are purified and phosphorylated by T4 polynucleotide kinase, PNK (E), then annealed to 3’ primer and the overhand is filled in using Klenow enzyme (F). After ligation with the 5’ duplex the selected sequences are PCR amplified for next round of selection. This protocol follows that described by Wen and Gray [
18].
Figure 3.
A schematic procedure for primer-free genomic SELEX. The genomic library is shown as dsDNA, at the conjunction between 5’ primer and genomic insert a ribose is indicated as 2’-OH, at the 3’ end the Fok I recognition site is indicated as lower case and its cutting sites are indicated by the red arrowheads. The starting library is generated by Fok I digestion followed by alkaline hydrolysis (A), after selection (B), it is annealed to dsDNA library (C) to synthesize the 3’ tail and fixed sequences (D). The extended products are purified and phosphorylated by T4 polynucleotide kinase, PNK (E), then annealed to 3’ primer and the overhand is filled in using Klenow enzyme (F). After ligation with the 5’ duplex the selected sequences are PCR amplified for next round of selection. This protocol follows that described by Wen and Gray [
18].
A bacteriophage fd genomic library was generated by introducing a restriction site in the 3' end and a ribose linkage in the 5' end primer-annealing regions, which were removed by enzymatic and alkaline treatments respectively (
Figure 3A). The tail regions, which usually contained a few point mutations due to the library construction, were also concomitantly removed by this procedure. The starting material, finally trimmed to ~57 nt and containing only genomic sequences, was used for the selection reaction with the target protein, g5p (
Figure 3B). To regenerate primer-annealing sites, the selected sequences were hybridized to the unselected dsDNA templates (
Figure 3C), and the hybridized inserts were then elongated by a thermo-stable DNA polymerase to synthesize the 3' primer-annealing sequence (
Figure 3D). The elongated products were separated from the templates on a denaturing gel (
Figure 3E), and the purified products were phosphorylated at the 5’ end by T4 polynucleotide kinase and then converted to blunt end duplexes by Klenow enzyme (
Figure 3F) before ligation of a 5’ end primer duplex (
Figure 3G). Finally, the selected sequences were amplified by PCR for the next round of selection (
Figure 3H). After four rounds of selection, a major group of sequences was identified and the major sequence, 57 nt, was tested in comparison with a downstream sequence as a control. It showed a ~23-fold higher binding, which demonstrated that the selected genomic sequences had higher affinity for g5p binding than an unrelated neighboring sequence.
This protocol relies on minimal diversity of the library for successful hybridization. Libraries or selection pools that are more complex than the fd genomic library have to be tested for efficiency of hybridization at this step. The fd genomic DNA is only 6408 nt in length, and it is theoretically represented by about the same number of unique sequences in the library. If a more complex starting library is used, a few initial rounds of regular or other types of SELEX could be performed to reduce the diversity of the selection pool, similar to the approach used in the primer-switching SELEX [
16], which would help decrease the chance of preferential selection of sequences with repeated elements, before the primer-free protocol is applied.
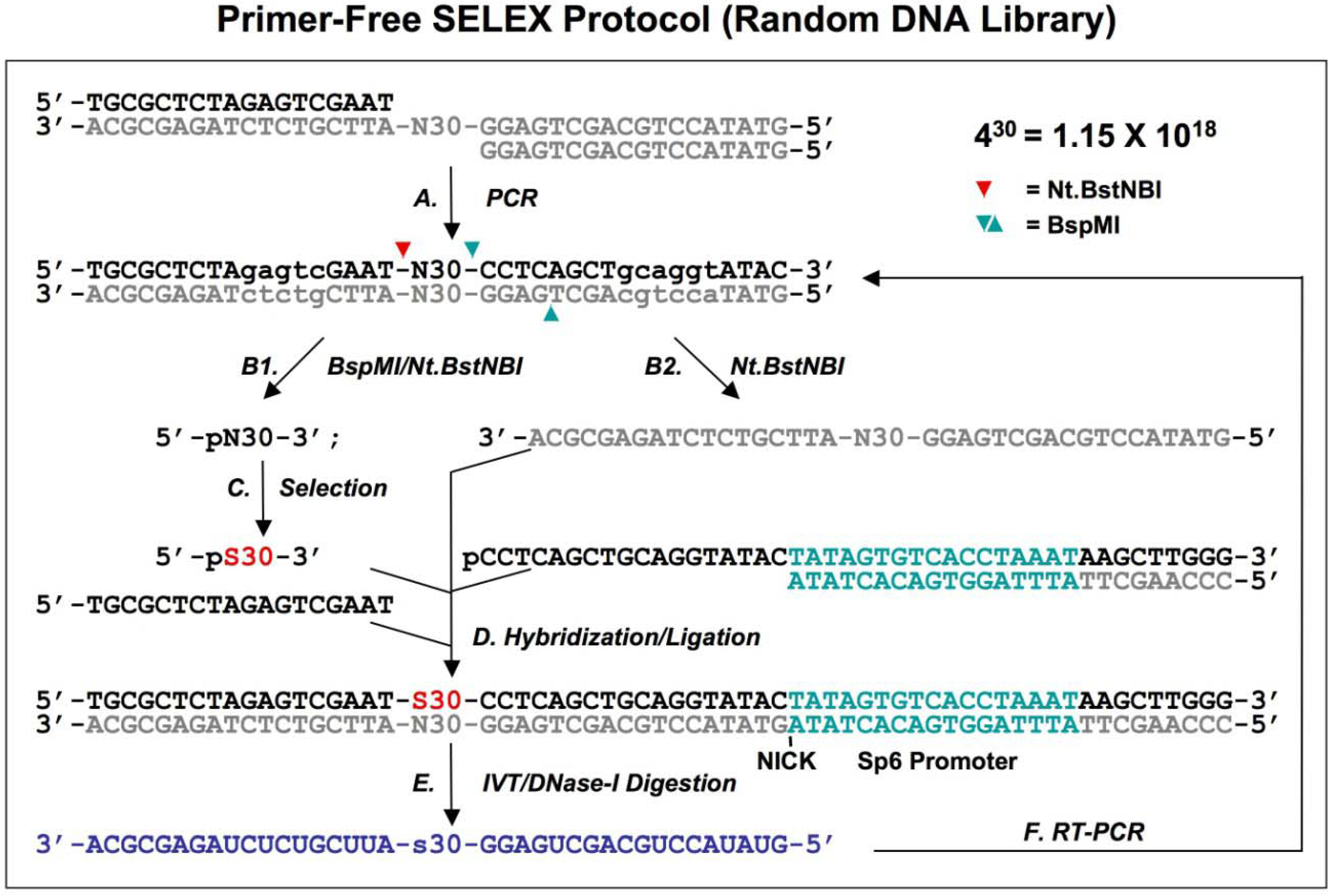
3. Primer-free 2’O-Methyl random RNA fishing SELEX
Most
in vitro selections are performed with unmodified RNA or DNA sequences, leading to aptamers of high affinity and specificity but with relatively short lifetimes
in vivo. Only a limited number of modified triphosphate nucleotides conferring nuclease resistance to the oligomer can be incorporated by polymerases. This encourages the development of alternative methods for the identification of nuclease-resistant aptamers. Boiziau and Toulme described such a “fishing” SELEX method [
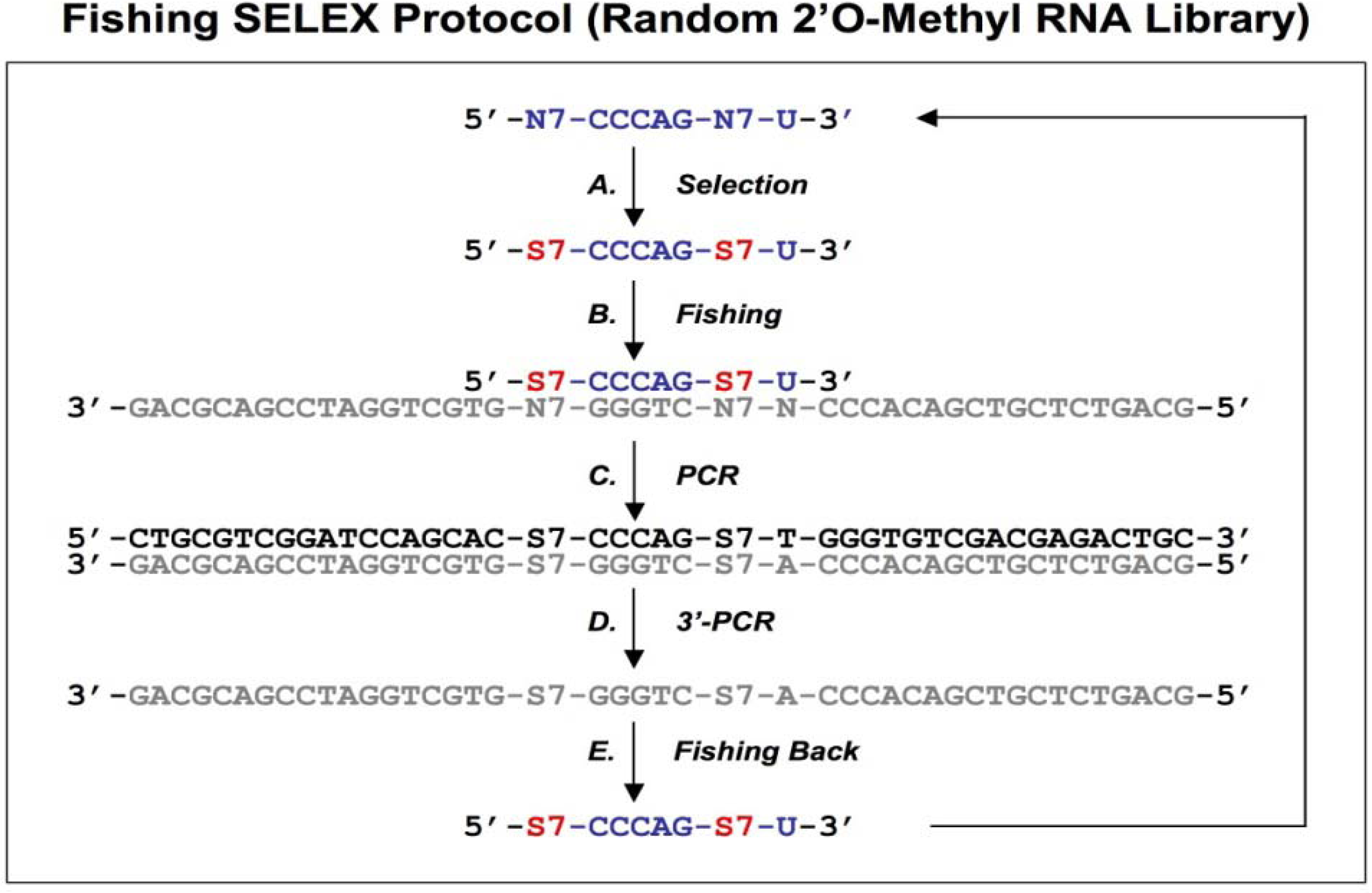
19]. After selection with a random 2’O-methyl RNA library against the TAR-RNA of HIV-1 (
Figure 4A), the complementary DNA sequences were fished out of a randomized DNA library by Watson-Crick hybridization (
Figure 4B). The DNA-fished sequences were PCR amplified as dsDNAs (
Figure 4C) and ssDNAs (
Figure 4D). The ssDNAs were used to fish back the chemically modified candidates from the initial library (
Figure 4E). This procedure allowed an indirect amplification of the selected candidates, and the enriched pool of 2’O-Methyl RNA was then used for the next round of selection.
Fifteen selected sequences were analyzed after six rounds of selection. No clear consensus emerged at this point of the selection although a non-statistical repartition of nucleotides was observed. Thus, this selection was still not sufficient, and no follow-up data were reported, suggesting limitations to the approach. Since a random library containing n nucleotides generates a 4n diversity, this method is limited to very short random regions. In this case, a 14 nt randomized library led to a theoretical population of 2.7 × 108 different molecules, which is over a million times smaller than standard starting pools of 1014-1015 molecules.
Figure 4.
Diagram of fishing SELEX using a random 2’O-Methyl RNA library. A chemically synthesized random 2’O-Methyl RNA library (indicated in blue) is incubated with target (A), the target bound sequences allow the fishing of the complementary DNA oligonucleotides (B). The fished DNA templates are amplified by regular PCR (C), and then the bottom strand sequences (indicated in gray) are synthesized by PCR using 3’ primers only (D). The amplified bottom strand sequences allow the fishing-back of the 2’O-Methyl RNA sequences, which are used for next round of selection. This protocol is from Boiziau and Toulme [
19].
Figure 4.
Diagram of fishing SELEX using a random 2’O-Methyl RNA library. A chemically synthesized random 2’O-Methyl RNA library (indicated in blue) is incubated with target (A), the target bound sequences allow the fishing of the complementary DNA oligonucleotides (B). The fished DNA templates are amplified by regular PCR (C), and then the bottom strand sequences (indicated in gray) are synthesized by PCR using 3’ primers only (D). The amplified bottom strand sequences allow the fishing-back of the 2’O-Methyl RNA sequences, which are used for next round of selection. This protocol is from Boiziau and Toulme [
19].
As a potential way to bypass these limitations, one option would be to perform a few rounds of selection with unmodified RNA pools; once some of convergent positions are identified, the chemically modified library could be synthesized to contain the convergent (fixed) nucleotide positions separated by a few short random regions. This restricted 2’O-Methyl RNA library might prove useful for further, more efficient selections.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








