Results and Discussion
Previous reported procedures [
8,
9,
10], were used for the first reaction, where
p-tert-butylthia-calixarene
1 was reacted with diethyl bromomalonate in refluxing acetone in the presence of K
2CO
3 as catalyst. Surprisingly, an unprecedented selective synthesis of monosubstituted
p-tert-butylthiacalixarene
2 containing one ethoxycarbonyl fragment (in 60 % yield) and the reported compound
3 [
11] resulted (
Scheme 1). When the reaction was carried out using Cs
2CO
3 as a base catalyst instead of K
2CO
3, a similar result was obtained, with a slight change in the yield of
2 (which increased to 70%). Different spectroscopic analysis (IR, 600 MHz
1H-NMR, and MALDI-TOF MS) were used to identify the structure of
p-tert-butylthiacalixarene
2. The
1H-NMR spectrum exhibits three Bu
t group singlets with a relative ratio of 1:1:2, two doublets and two singlets in the aromatic region, one triplet for the CH
3 group, one quartet for the OCH
2 group and two singlets of OH group with a relative ratio 2:1, respectively. MALDI-TOF MS shows an m/z peak at 832 (M+K). IR shows bands at 1758 and 3380 cm
-1 for the CO and OH groups, respectively.
Since the ethoxycarbonyl fragment in
p-tert-butylthiacalixarene
2 could be introduced to
p-tert-butylthiacalixarene
1, through the reaction with ethyl chloroformate, and to investigate the selectivity of the monosubstituted
p-tert-butylthiacalixarene
2, ethyl chloroformate was reacted with
p-tert-butylthiacalixarene
1 in the presence of K
2CO
3 in acetone by using the same reaction conditions employed with diethyl bromomalonate. The tetrasubstituted
p-tert-butylthiacalixarene
4, was obtained in 80 % yield (
Scheme 1; separated by
partial cone conformer) which is in agreement with the literature [
8,
9,
10]. The structure of
4 was characterized using IR,
1H-NMR and MALDI-TOF-MS spectroscopy. The above selective synthesis of the monosubstitued thiacalixarene
2 may be explained by the generation of the ethoxycarbonyl fragment throughout the decomposition of the diethyl bromomalonate in the basic medium or by the attack of the phenolate-metal ion pairs at one of the carbonyl groups of the diethyl bromomalonate and metal ion template effect (Cs and K). This implies that the selective monosubstitution can be attributed to the reaction of thiacalixarene
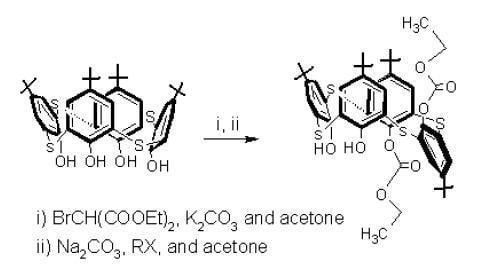
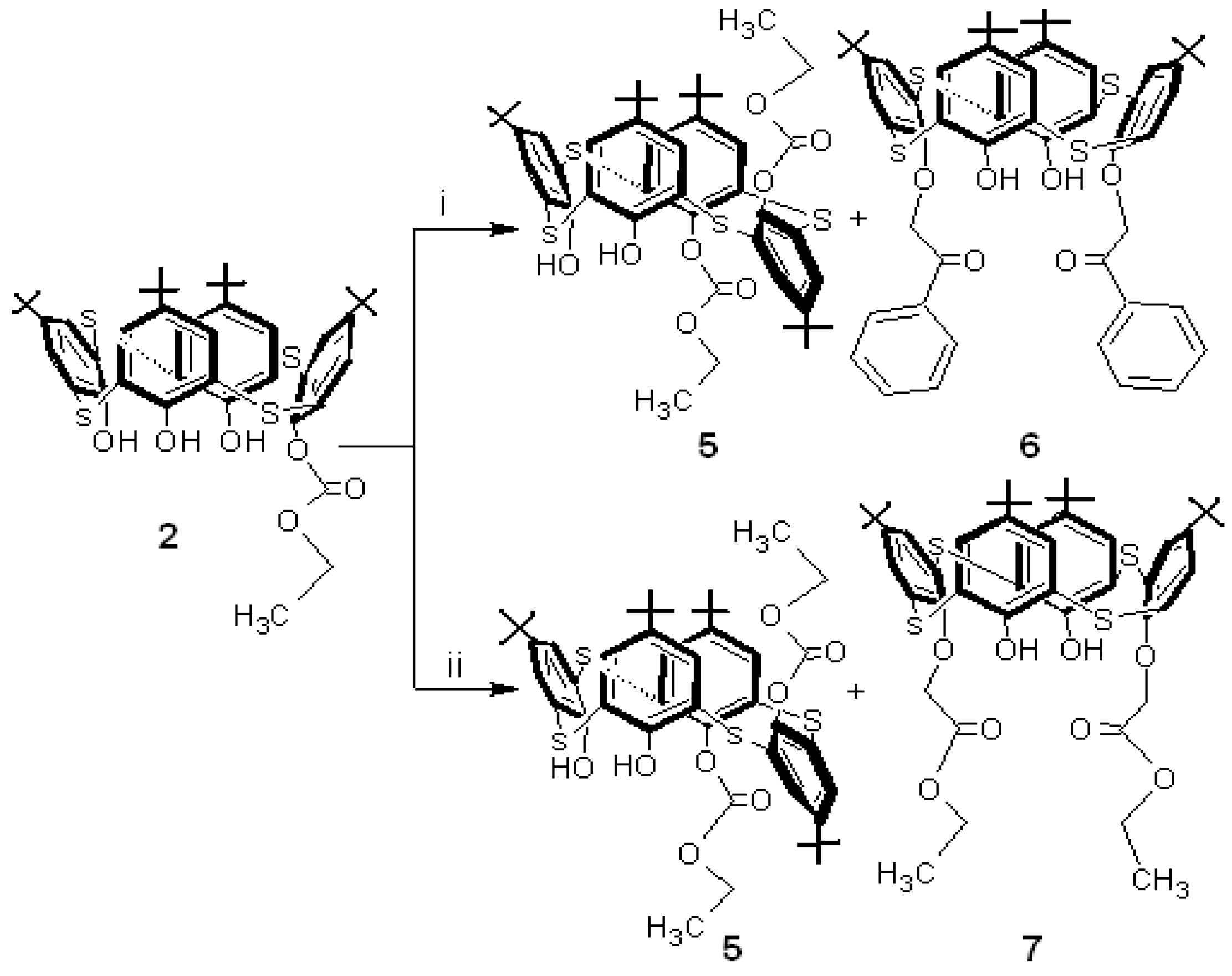
1 with diethyl bromomalonate. For the preparation of asymmetrical
p-tert-butylthiacalixarenes, introducing four different groups at the lower rims, monosubstituted thiacalixarene
2 was used for the second step in this study – the reaction of
2 with phenacyl bromide in the presence of Na
2CO
3 in acetone (
Scheme 2).
The reaction gave
anti-1,2-disubstituted
p-tert-butylthiacalixarene
5 [
12] in 40% yield (dissymmetrical thiacalixarene has effective C
2 symmetry) and the reported disubstituted
p-tert-butylthiacalixarene
6 [
13].
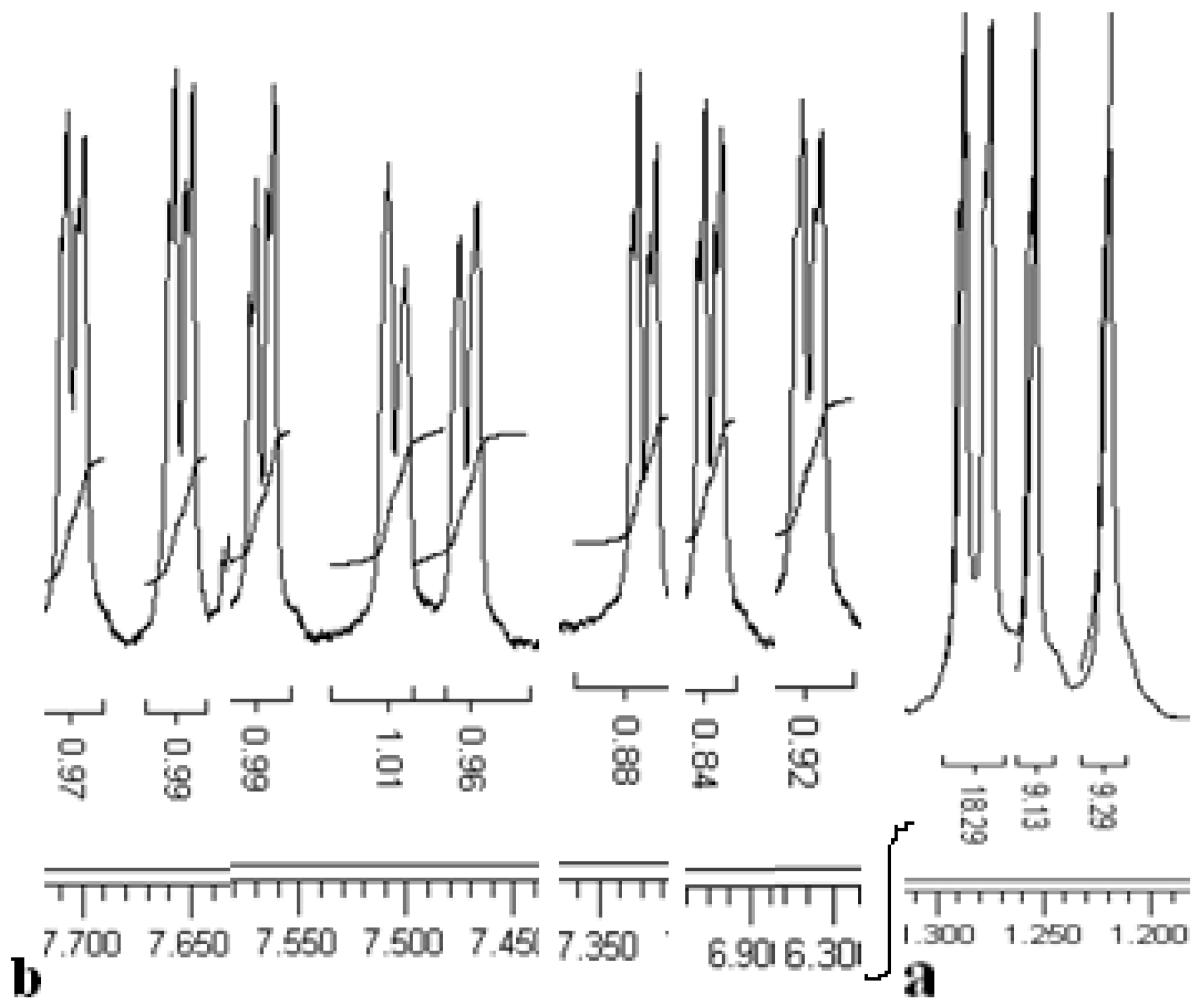
The
1H-NMR spectrum of compound
5 (
Figure 1) shows asymmetrical signals for all protons which have been used for the structure determination: four Bu
t group singlets, eight doublets with characteristic aromatic proton coupling constants (
J ~ 2.3 to 2.4 Hz), two OH group singlets, two CH
3 group triplets, a quartet for the CH
2, shifted upfield by the aromatic rings and one CH
2 group multiplet in between two different groups (OH and Ar groups). To confirm the production of
p-tert-butylthiacalixarene
5,
p-tert-butylthiacalixarene
2 was reacted with another alkylation reagent, ethyl bromoacetate and
p-tert-butylthiacalixarene
5 was formed along with the reported
p-tert-butyl-thiacalixarene
7 [
14] (
Scheme 2).
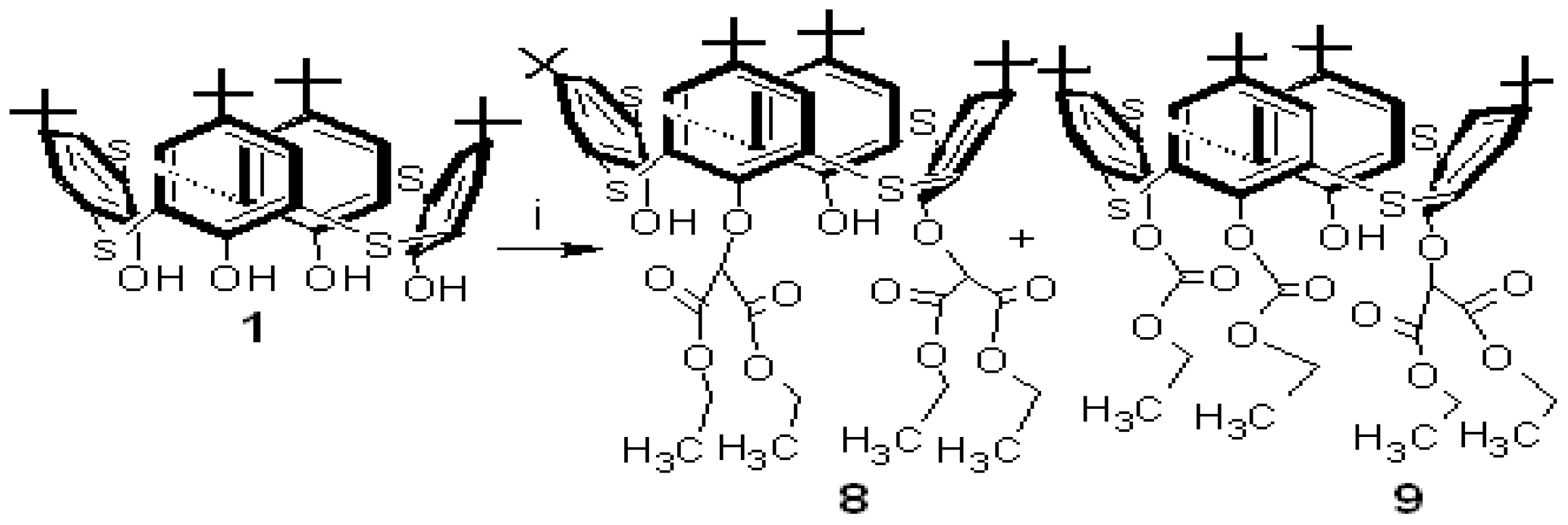
The reaction of
p-tert-butylthiacalixarene
1 with diethyl bromomalonate in the presence of Na
2CO
3 in acetone gave two new
p-tert-butylthiacalixarenes,
8 and
9, in 30% and 40% yields, respectively (
Scheme 3).
Both p-tert-butylthiacalixarenes 8 and 9 were separated from the carbonate and the acetone layer, respectively. IR, MALDI-TOF-MS and 1H-NMR spectroscopic methods were used for determination of the their structures.
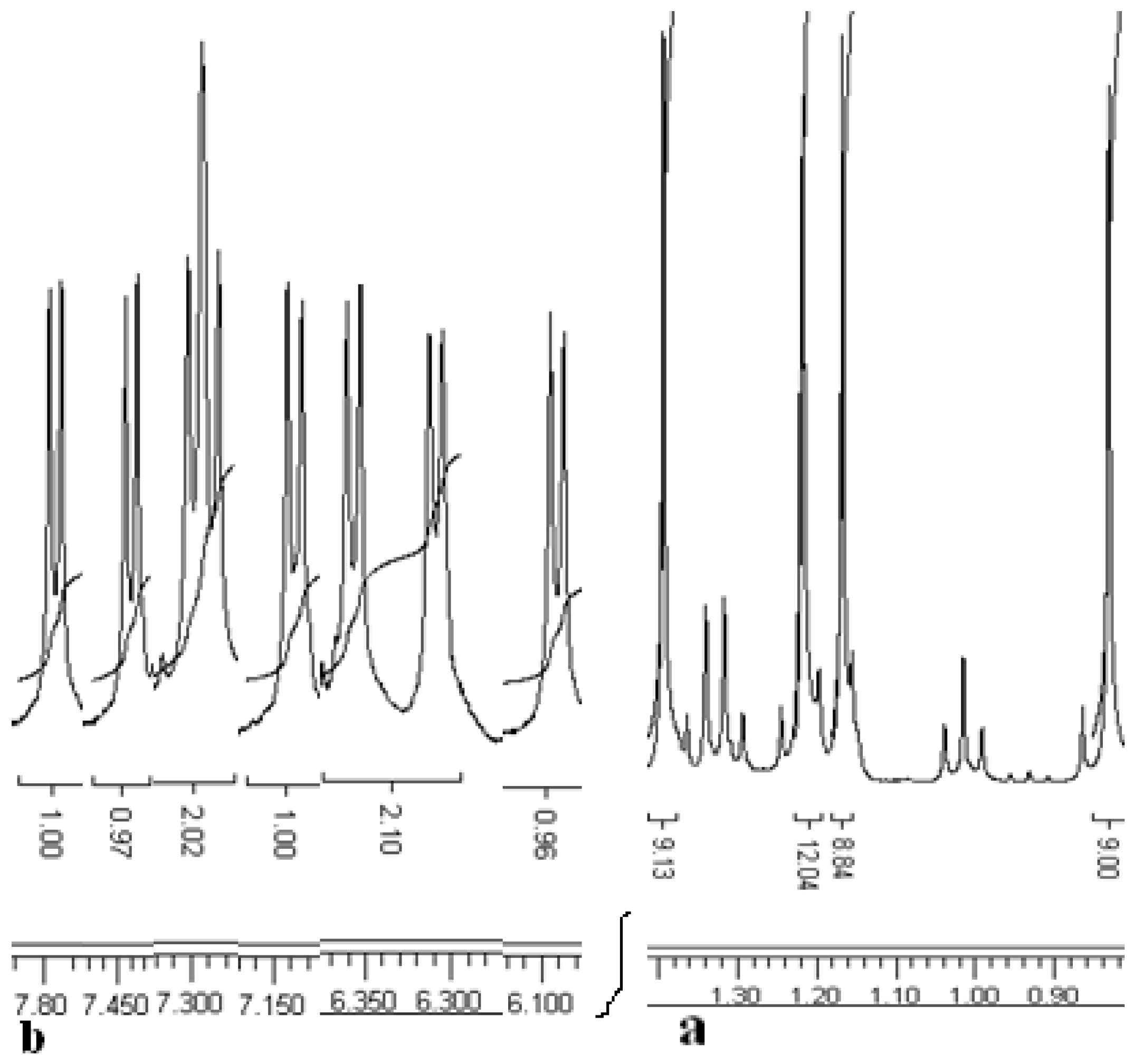
The
1H-NMR spectrum of compound
8 exhibits two Bu
t group singlets, four multiplets for the methylene groups and four doublets with characteristic meta coupling in the aromatic region (
J ~ 2.4 Hz). The
1H-NMR spectrum of asymmetrical thiacalixarene
9 shows asymmetrical signals for all the types of protons, for instance, four singlets for the Bu
t groups, four CH
3 group triplets, four CH
2 group multiplets and eight doublets with characteristic coupling constants (
J ~ 2.3 to 2.8 Hz) in the aromatic region (
Figure 2).
Experimental
General
All NMR spectra were recorded on a Bruker DRX600 NMR spectrometer equipped with a triple-gradient TXI (H/C/N) probe operating at a magnetic field strength of 14.1 T., as well as on a Varian Mercury VX-300 NMR spectrometer. All melting points were determined on a Koffler melting point apparatus. IR spectra were obtained on a Nicolet 710 FT-IR spectrometer. Mass spectra were recorded on a MALDI-TOF MS REFLEX III (Bruker-Daltonics, Germany).
Synthesis of thiacalixarene 2
To a suspention of thiacalixarene 1 (2 g, 2.77 mmol) and anhydrous potassium carbonate (6.16 g, 44.32 mmol) and/or cesium carbonate (7.21 g, 22.16 mmol) in dry acetone (100 mL) diethylbromomalonate (8.2 mL, 44.32 mmol) was added. The mixture was refluxed for a week, then it was filtered and separated into two layers: carbonate and acetone. Compound 2 was separated from the carbonate layer after extraction by CH2Cl2 and obtained as a white solid. Compound 3 was separated from the acetone layer.
5,11,17,23-Tetra-tert-butyl-25-[(ethoxycarbonyl)oxy]-26,27,28-trihydroxy-2,8,14,20-tetrathiacalix[4]- arene (cone) (2): Yield. 60-70%; Mp: 218 °C; IR (cm-1): 1758 (CO), 3380 (OH); MS m/z 832 (M+K); 1H-NMR (TMS, 300 MHz, CDCl3) δ 0.76 (9H, s, But ), 1.16 (9H, s, But), 1.28 (18H, s, But), 1.36 (3H, t, J =7.1, CH3), 4.46 (2H, q, J = 7.1, CH2), 7.04 (2H, s, ArH), 7.48 (2H, s, ArH), 7.60 (2H, d, J = 2.4, ArH), 7.73 (2H, d, J = 2.4, ArH), 8.20 (2H, s, OH), 9.10 (1H, s, OH).
Synthesis of thiacalixarene 4
The above method was used for the preparation of 4, obtained as a white solid.
5,11,17,23-Tetra-tert-butyl-25,26,27,28-tetrakis[(ethoxycarbonyl)oxy]-2,8,14,20-tetrathiacalix[4]-arene (partial cone) (4): Yield: 80%; Mp: 330 °C; IR (cm-1): 1756 (CO); MS m/z 1072 (MH+); 1H-NMR (TMS, 300 MHz, CDCl3) δ 1.09 (18H, s, But ), 1.38 (9H, s, But), 1.47 (9H, s, But), 1.33-1.50 (12H, m, CH3), 4.19 (2H, q, J = 7.0, CH2), 4.26-4.48 (6H, m, CH2), 7.18 (2H, d, J = 2.4, ArH), 7.44 (2H, s, ArH), 7.45 (2H, d, J = 2.4, ArH), 7.78 (2H, s, ArH).
Synthesis of thiacalixarenes 5, 6 and 7
Thiacalixarene 2 (0.6 g, 0.75 mmol) and Na2CO3 (0.1 g, 0.94 mmol) were suspended in acetone (50 mL) and phenacyl bromide (0.15 g, 0.75 mmol) or ethyl bromoacetate (0.1 mL, 0.9 mmol) were added. The mixture was refluxed for 24 hours. The reaction mixture was concentrated almost to dryness and the solid residue was separated by column chromatography using a 50/50 n-hexane-CH2Cl2 mixture as eluent, to separate the thiacalixarenes 5 and 6 from the reaction with phenacyl bromide; or thiacalixarenes 5 and 7 from the reaction with ethyl bromoacetate.
5,11,17,23-Tetra-tert-butyl-25,26-bis[(ethoxycarbonyl)oxy]-27,28-dihydroxy-2,8,14,20-tetrathiacalix-[4]arene (anti-1,2-alternate) (5) : Yield: 40%; yellow solid; Mp: 310 °C; IR (cm-1): 1754 (CO); MS m/z 889 (M+Na); 1H-NMR (TMS, 300 MHz, CDCl3) δ 1.07-1.17 (6H, m, CH3), 1.22 (9H, s, But), 1.26 (9H, s, But), 1.28 (9H, s, But), 1.29 (9H, s, But), 3.7 (2H, q, J = 3.7, CH2), 4.14-4.34 (2H, m, CH2), 6.31 (2H, d, J = 2.3, ArH), 6.92 (2H, q, J = 2.3, ArH), 7.33 (2H, d, J = 2.4, ArH), 7.39 (1H, s, OH), 7.47 (2H, d, J = 2.4, ArH), 7.51 (2H, d, J = 2.4, ArH), 7.57 (2H, d, J = 2.4, ArH), 7.66 (2H, d, J = 2.4, ArH), 7.71 (2H, d, J = 2.4, ArH), 7.81 (1H, s, OH).
Synthesis of thiacalixarenes 8 and 9
The same procedure for the preparation of thiacalixarene 2 was used. Thiacalixarene 8 was separated from the carbonate layer and thiacalixarene 9 was separated from the acetone layer.
5,11,17,23-Tetra-tert-butyl-25,26-bis[(1,1-diethoxycarbonyl)methoxy]-27,28-dihydroxy-2,8,14,20-tetrathiacalix[4]arene (cone) (8): Yellow solid; Mp: 330 °C; Yield: 30%; IR (cm-1): 1751 (CO); MS m/z 1061 (M+Na); 1H-NMR (TMS, 300 MHz, CDCl3) δ 0.96 (6H, t, J = 7.1, CH3), 1.18 (6H, t, J = 7.1, CH3), 1.21 (18H, s, But ), 1.23 (18H, s, But), 3.72 (2H, m, CH2), 3.79 (2H, m, CH2), 3.93-4.04 (6H, m, CH2 + OCH), 6.05 (2H, q, J = 2.4, ArH), 6.87 (2H, d, J = 2.4, ArH), 7.27 (2H, s, OH), 7.44 (2H, d, , J = 2.4, ArH), 7.65 (2H, d, J = 2.4, ArH).
5,11,17,23-Tetra-tert-butyl-25-[(1,1-diethoxycarbonyl)methoxy]-26,28-bis[(ethoxycarbonyl)oxy-26-hydroxy-2,8,14,20-tetrathiacalix[4]arene (cone) (9): Yellow solid; Yield: 40%; Mp: 275 °C; IR (cm-1): 1758 (CO); MS m/z 1047 (M+Na); 1-NMR (TMS, 300 MHz, CDCl3) δ = 0.84 (9H, s, But ), 1.01 (6H, t, J = 7.1, CH3) 1.17 (9H, s, But) 1.22 (9H, s, But), 1.22 (6H, t, J = 7.1, CH3), 1.33 (12H, m, CH3), 1.4 (9H, s, But), 3.98- 4.50 (10H, m, CH2 + OCH, OH), 6.10 (2H, d, J = 2.3, ArH), 6.31 (2H, q, J = 2.3, ArH), 6.36 (2H, d, J = 2.3, ArH), 7.14 (2H, d, J = 2.5, ArH), 7.29 (2H, d, J = 2.8, ArH), 7.30 (2H, d, J = 2.8, ArH), 7.44 (2H, d, J = 2.5, ArH), 7.80 (2H, d, J = 2.5, ArH).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




