A Facile Route to C2-Substituted Imidazolium Ionic Liquids

Abstract
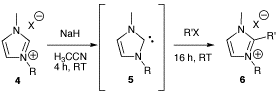
:
Introduction
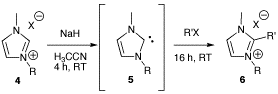
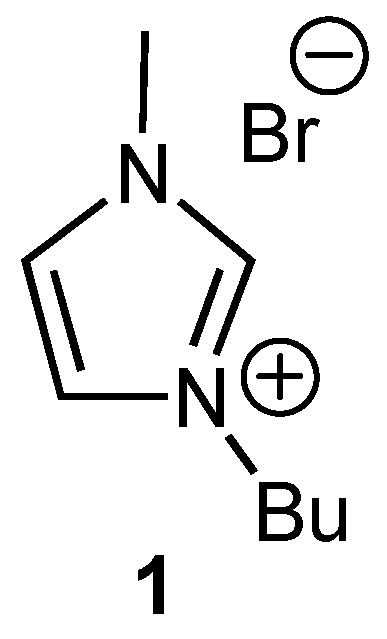
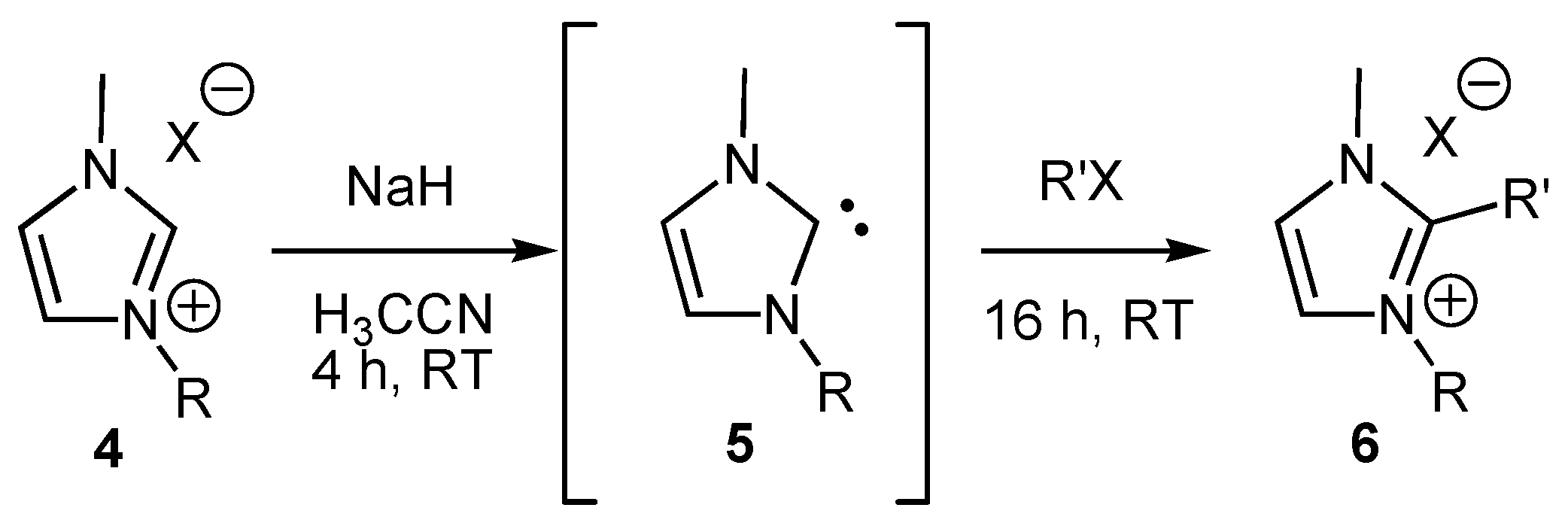
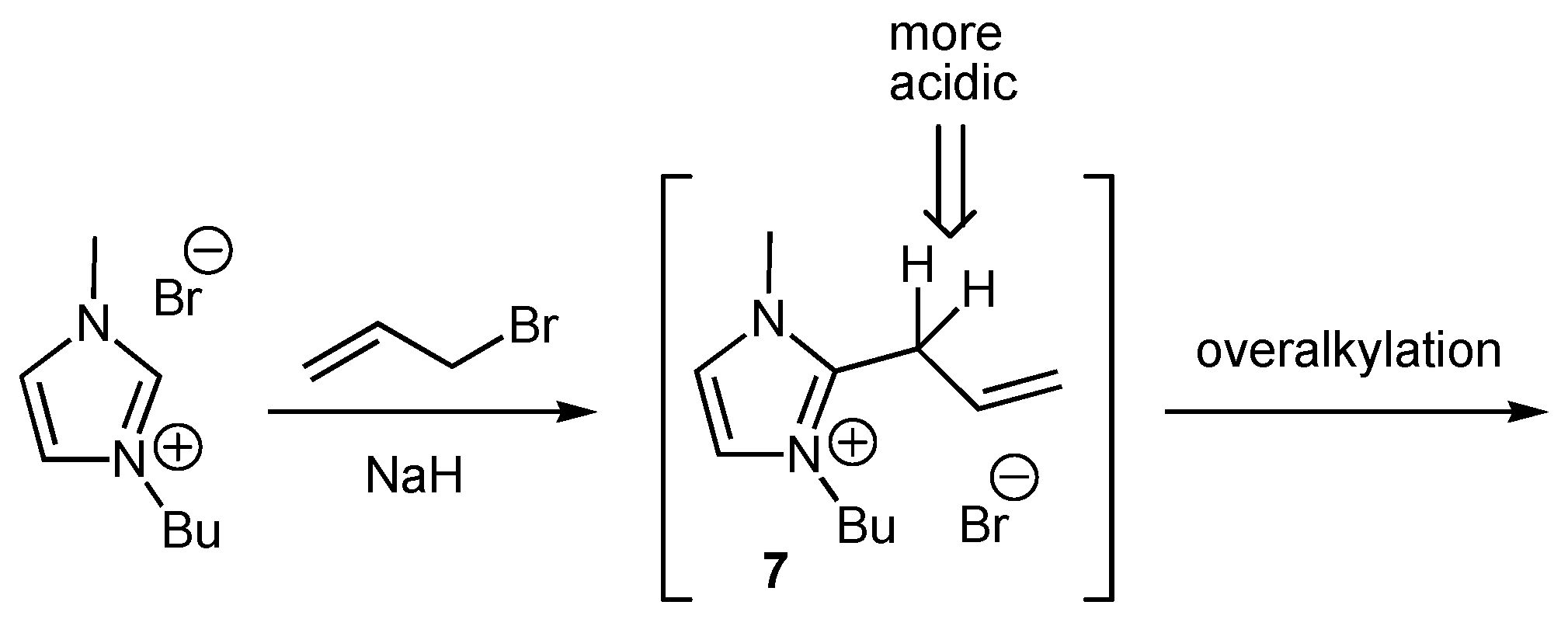
2. Results and Discussion
Experimental
General
General procedure for the alkylation of 1-butyl-3-methylimidazolium bromide (BMIM Br) with alkyl chlorides
Synthesis of 1-butyl-2-ethyl-3-methylimidazolium bromide (BEMIM Br)
Synthesis of 2-butyl-1-ethyl-3-methylimidazolium iodide (EBMIM I)
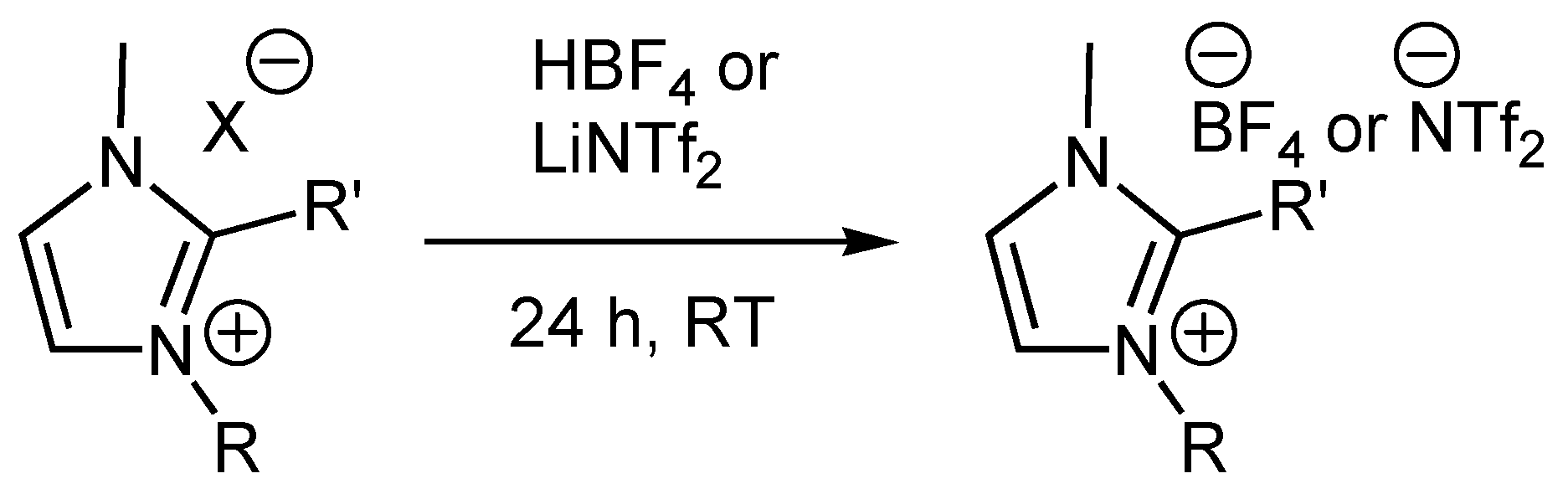
Representative Anion Metathesis to afford Tetrafluoroborate Salts
Representative Anion Metathesis to afford Triflimide Salts
Conclusions
Acknowledgements
References and Notes
- Welton, T. Room-Temperature Ionic Liquids: Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, M. Eyes on Ionic Liquids. C&E News 2000, 78, 37–50. [Google Scholar]
- Wasserscheid, P.; Welton, T. (Eds.) Ionic Liquids in Synthesis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Handy, S.T. Room Temperature Ionic Liquids: Different Classes and Physical Properties. Curr. Org. Chem. 2005, 9, 959–988. [Google Scholar] [CrossRef]
- Wilkes, J.S. Properties of ionic liquid solvents for catalysis. J. Mol. Cat. A Chem. 2004, 214, 11–17. [Google Scholar] [CrossRef]
- Ennis, E.; Handy, S.T. The Chemistry of the C2 Position of Imidazolium Room Temperature Ionic Liquids. Curr. Org. Chem. 2007, 4, 381–389. [Google Scholar] [CrossRef]
- Chowdhury, S.; Mohan, R.S.; Scott, J.L. Reactivity of Ionic Liquids. Tetrahedron 2007, 63, 2363–2389. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Emme, I.; Mereu, A. Unexpected side reaction of imidazolium-based ionic liquids in the base-catalysed Baylis-Hillman reaction. Chem. Commun. 2002, 1612–1613. [Google Scholar] [CrossRef]
- Handy, S.T. Grignard Reactions in Imidazolium Ionic Liquids. J. Org. Chem. 2006, 71, 4659–4652. [Google Scholar] [CrossRef] [PubMed]
- Begtrub, M. Introduction of Substituents into 5-membered Aza-Heteroaromatics. Bull. Soc. Chim. Belg. 1988, 97, 573–597. [Google Scholar] [CrossRef]
- Alder, R.W.; Allen, P.R.; Williams, S.J. Stable Carbenes as Strong Bases. J. Chem. Soc., Chem. Commun. 1995, 1267–1268. [Google Scholar] [CrossRef]
- Arduengo, A.J.; Rasika Dias, H.V.; Harlow, R.L.; Kline, M. Electronic Stabilization of Nucleophilic Carbenes. J. Am. Chem. Soc. 1992, 114, 5530–5534. [Google Scholar] [CrossRef]
- Bourissou, D.; Guerret, O.; Gabbai, F.P.; Bertrand, G. Stable Carbenes. Chem. Rev. 2000, 100, 39–92. [Google Scholar] [CrossRef] [PubMed]
- The identity of the halide in these salts has not yet been rigorously determined. It is speculated to be a bromide, since reactions of bmim bromide with either alkyl bromides or alkyl chlorides give materials with identical 1H-NMR spectra, whereas the product of alkylation of bmim chloride with an alkyl chloride gives a material with slightly different chemical shifts for the two imidazolium signals. As a result, it appears that the bromide remains the counterion in these alkylation reactions, even when it could be mixed with a chloride. Further work on confirming this observation is underway and will reported in due course.
- Bonhote, P.; Dias, A.P.; Papageorgiou, N. Hydrophobic, Highly Conductive Ambient-Temperature Molten Salts. Inorg. Chem. 1996, 35, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J. S.; Zaworotko, M. J. Air and Water Stable 1-ethyl-3-methyl imidazolium based Ionic Liquids. J. Chem. Soc., Chem. Commun. 1992, 965–967. [Google Scholar] [CrossRef]
- Xu, D.-Q.; Liu, B.-Y.; Luo, S.-P.; Xu, Z.-Y. A Novel Eco-friendly Method for the Preparation of Ionic Liquids. Synthesis 2003, 2626–2628. [Google Scholar] [CrossRef]
- Egashira, M.; Yamamoto, Y.; Fukutake, T.; Yoshimoto, N. A novel method for the preparation of imidazolium tetrafluoroborate ionic liquids. J. Fluor. Chem. 2006, 127, 1261–1264. [Google Scholar] [CrossRef]
- It should be noted that the water content of the samples used for the viscosity measurements is not known. All samples were dried in a uniform fashion at 100°C overnight under high vacuum. This procedure afforded materials that consistently gave viscosity measurements that were within a 5% range of the reported values.
- Huddleston, J.G.; Visser, A.E.; Reichert, W.M.; Willauer, H.D.; Broker, G.A.; Rogers, R.D. Characterization and comparison of hydrophilic and hydrophobic room temperature ionic liquids incorporating the imidazolium cation. Green Chem. 2001, 3, 156–164. [Google Scholar] [CrossRef]
Sample Availability: Small samples are available from the author. |






{kind=link}
{kind=link}
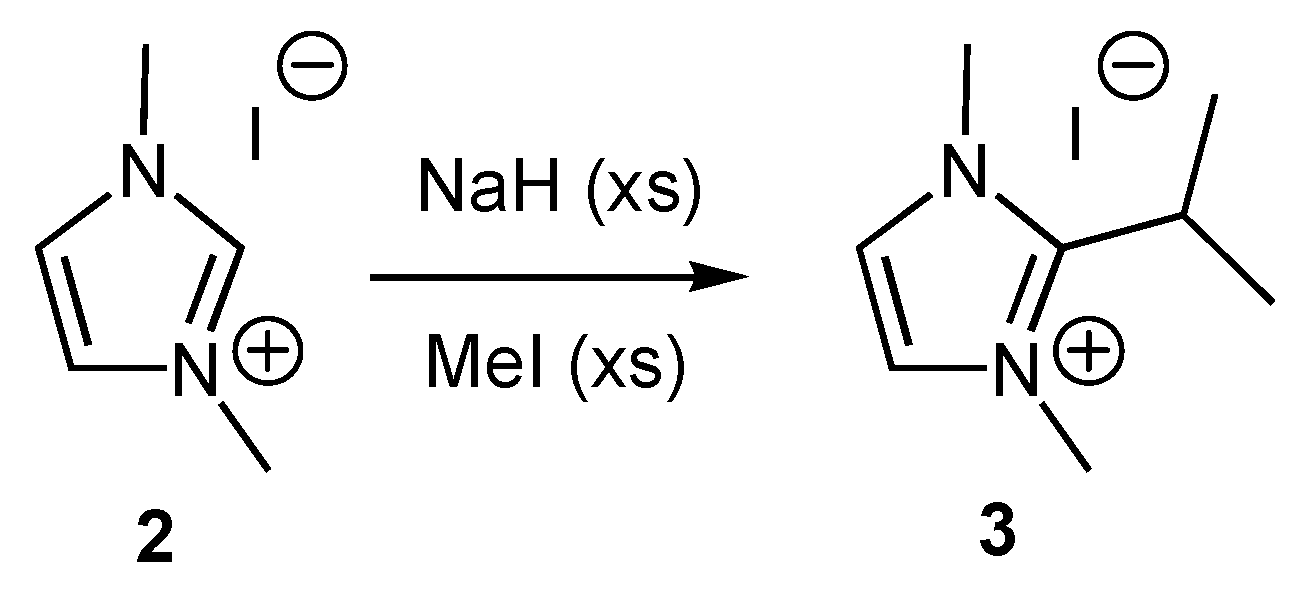{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Alkyl Halide | Equivalents | % yielda |
|---|---|---|---|
| 1 | Iodoethane | 5 | 99 |
| 2 | Bromoethane | 4 | 99 |
| 3 | Chlorobutane | 3 | 92 |
| 4 | Iodobutanea | 3 | (50) |
| 5 | Chlorohexane | 3 | 85 |
| 6 | Bromohexanea | 3 | (93) |
| 7 | Chloroheptane | 3 | 67 |
| 8 | Bromoheptanea | 3 | (70) |
| 9 | Chlorodecane | 3 | 85 |
| 10 | Chlorohexadecane | 3 | NAb |
| 11 | Bromobutanea | 3 | (96) |
| Entry | R | R’ | Anion | % Yield |
|---|---|---|---|---|
| 1 | Bu | Bu | BF4 | 53 |
| 2 | Bu | Hex | BF4 | 64 |
| 3 | Bu | Heptyl | BF4 | 54 |
| 4 | Bu | Decyl | BF4 | 56 |
| 5 | Bu | Bu | NTf2 | 71 |
| 6 | Et | Bu | BF4 | 44 |
| 7 | Et | Bu | NTf2 | 83 |
| Entry | R | R’ | Anion | MP | Visc. |
|---|---|---|---|---|---|
| 1 | Bu | Et | Br | 46-52 | NA |
| 2 | Bu | Bu | Br | L | 1,760 |
| 3 | Bu | Bu | BF4 | L | 400 |
| 4 | Bu | Bu | NTf2 | L | 224 |
| 5 | Bu | Hex | Br | 20ish | NA |
| 6 | Bu | Hex | BF4 | L | 1,170 |
| 7 | Bu | Heptyl | Br | 48-55 | NA |
| 8 | Bu | Heptyl | BF4 | L | 963 |
| 9 | Bu | Decyl | Br | 50-60 | NA |
| 10 | Bu | Decyl | BF4 | L | 380 |
| 11 | Et | Bu | I | 47-55 | NA |
| 12 | Et | Bu | BF4 | L | 220 |
| 13 | Et | Bu | NTf2 | L | 48.3 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ennis, E.; Handy, S.T. A Facile Route to C2-Substituted Imidazolium Ionic Liquids. Molecules 2009, 14, 2235-2245. https://doi.org/10.3390/molecules14062235
Ennis E, Handy ST. A Facile Route to C2-Substituted Imidazolium Ionic Liquids. Molecules. 2009; 14(6):2235-2245. https://doi.org/10.3390/molecules14062235
Chicago/Turabian StyleEnnis, Elliot, and Scott T. Handy. 2009. "A Facile Route to C2-Substituted Imidazolium Ionic Liquids" Molecules 14, no. 6: 2235-2245. https://doi.org/10.3390/molecules14062235
APA StyleEnnis, E., & Handy, S. T. (2009). A Facile Route to C2-Substituted Imidazolium Ionic Liquids. Molecules, 14(6), 2235-2245. https://doi.org/10.3390/molecules14062235