Synthesis and Antitumor Evaluation of Novel Bis-Triaziquone Derivatives

Abstract
:Introduction
Results and Discussion


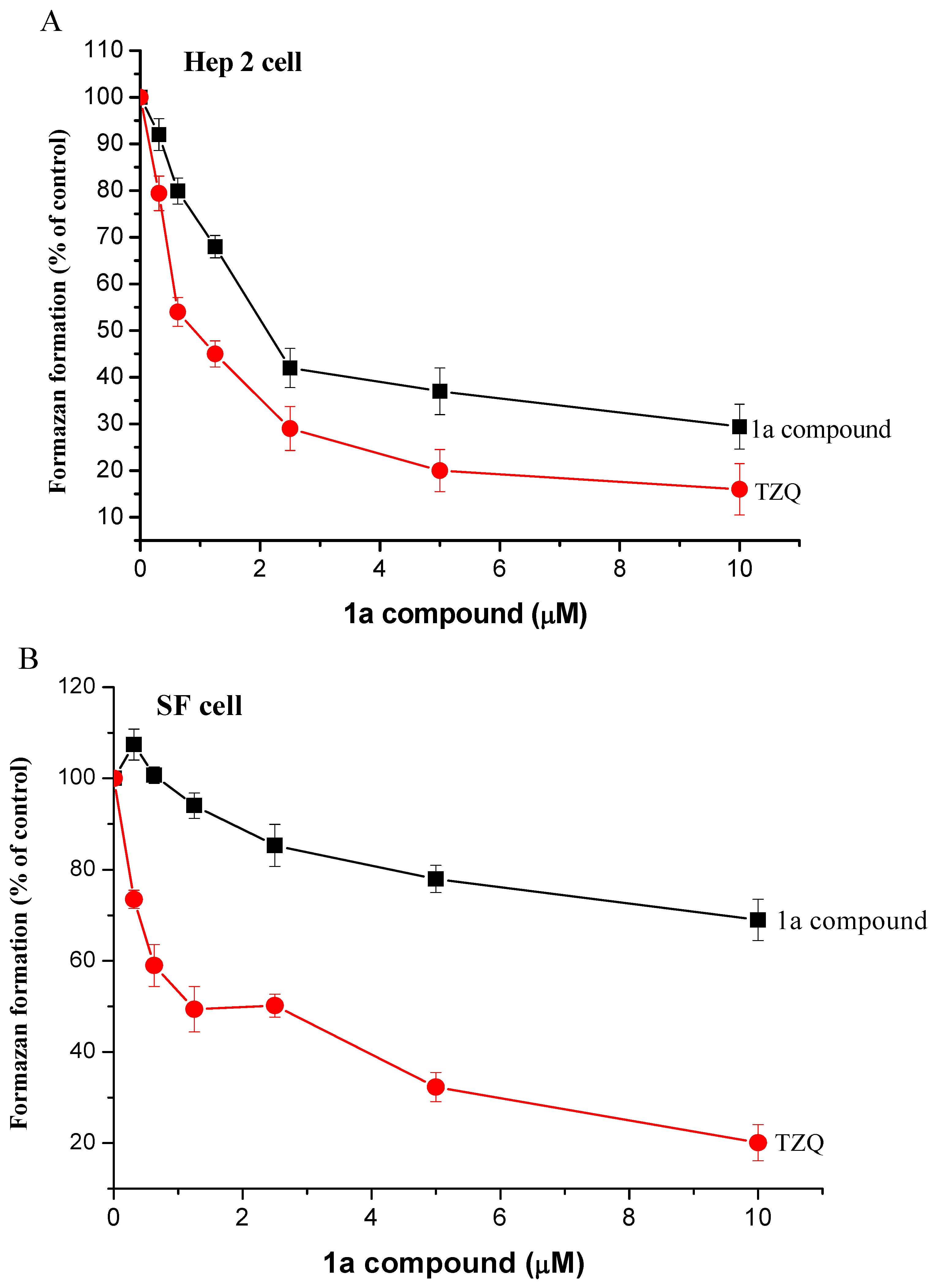{kind=link}
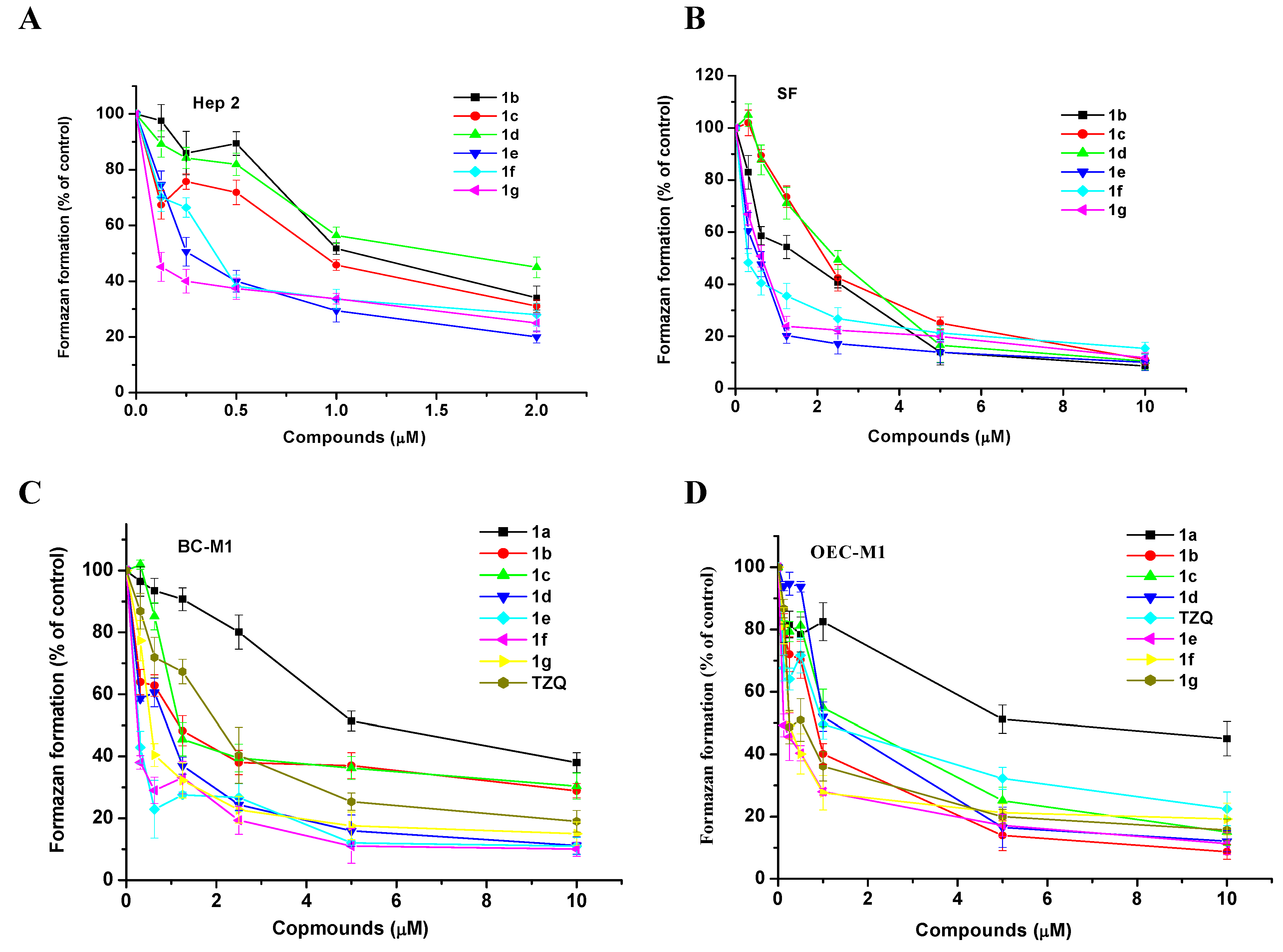{kind=link}
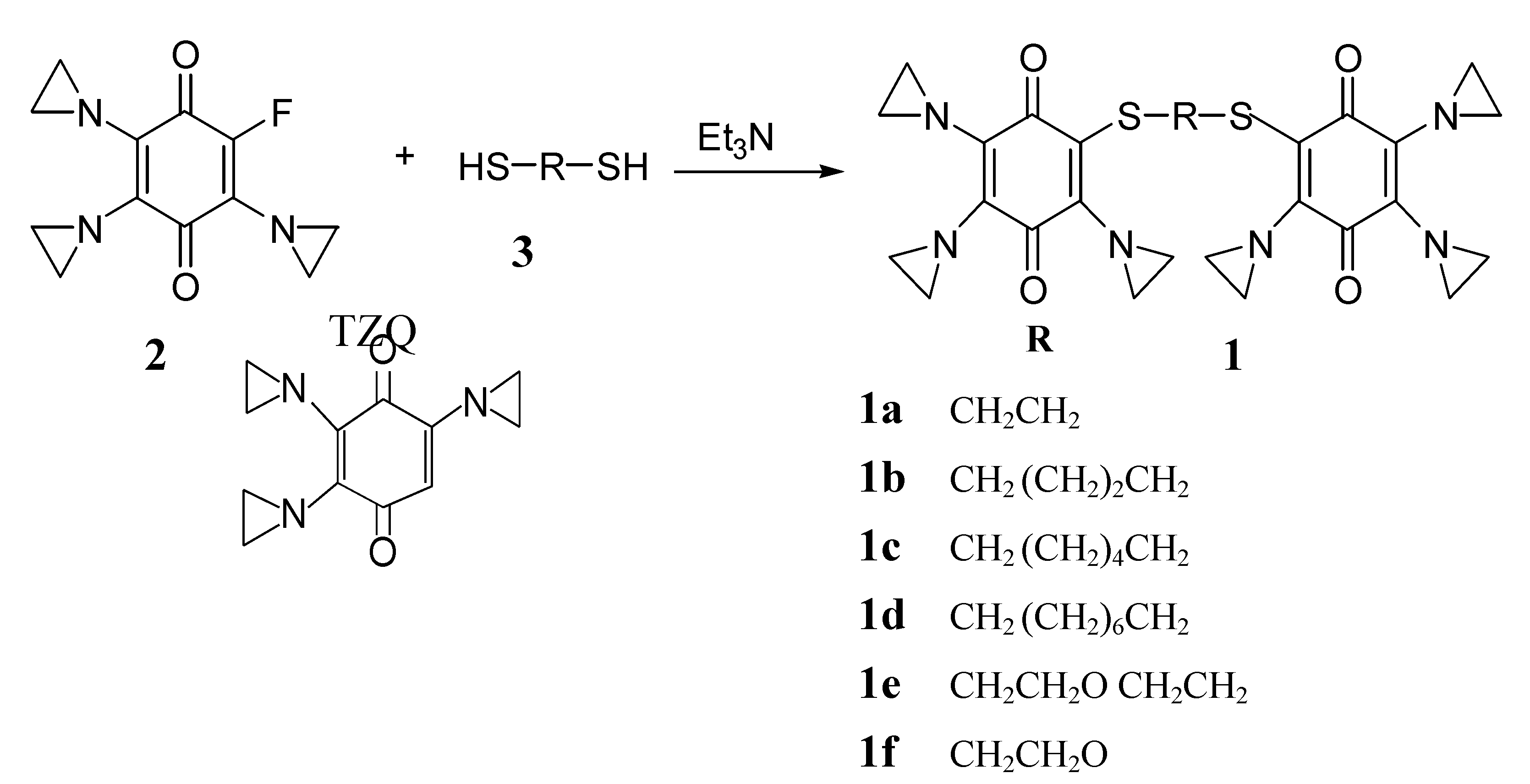{kind=link}
| Compound | Hep2 | OEC-M1 | BC-M1 | SF |
|---|---|---|---|---|
| 1a | 2.02±0.16 | 5.02±0.34 | 5.52±0.28 | > 10 |
| 1b | 1.02±0.04 | 0.85±0.05 | 1.25±0.16 | 2.12±0.12 |
| 1c | 0.88±0.06 | 1.52±0.12 | 1.07±0.09 | 2.35±0.13 |
| 1d | 1.82±0.14 | 1.14±0.07 | 0.89±0.14 | 2.48±0.14 |
| 1e | 0.25±0.08 | 0.11±0.04 | 0.21±0.06 | 0.63±0.07 |
| 1f | 0.42±0.03 | 0.21±0.03 | 0.21±0.04 | 0.32±0.04 |
| 1g | 0.11±0.02 | 0.22±0.02 | 0.53±0.10 | 0.63±0.08 |
| TZQ | 0.72±0.05 | 1.02±0.11 | 2.05±0.17 | 2.52±0.17 |


Experimental
General
General procedure for preparing bis-triazquones 1a-g
Cell culture
Cytotoxicity assay (MTT assay)
Conclusion
Acknowledgments
References and Notes
- Brown, J.M. Tumor microenvironment and the response to anticancer therapy. Cancer Biol. Ther. 2002, 1, 453–458. [Google Scholar] [CrossRef]
- Robert, G.B.; Stuart, T. Hypoxia: Targeting the tumour. Anticancer Agents Med. Chem. 2006, 6, 281–286. [Google Scholar] [CrossRef]
- Wouters, B.G.; Weppler, S.A.; Koritzinsky, M.; Landuyt, W.; Nuyts, S.; Theys, J.; Chiu, R.K.; Lambin, P. Hypoxia as a target for combined modality treatments. Eur. J. Cancer 2002, 38, 240–257. [Google Scholar] [CrossRef]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumor hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- McKeown, S.R.; Coweny, R.L.; Williamsy, K.J. Bioreductive Drugs: from Concept to Clinic. Clin. Oncol. 2007, 19, 427–442. [Google Scholar] [CrossRef]
- Phillips, R.M.; Burger, A.M.; Loadman, P.M.; Jarrett, C.M.; Swaine, D.J.; Fiebig, H.H. Predicting tumor responses to mitomycine C on the basis of DT-diaphorase activity or drug metabolism by tumorhomogenares: implications for enzyme-directed bioreductive drug development. Cancer Res. 2000, 60, 6384–6390. [Google Scholar]
- Kim, J.Y.; Kim, C.H.; Stratford, I.J.; Patterson, A.V.; Hendry, J.H. The bioreductive agents RH1 and gamma-irradition both cause G2/M cell cycle phase arrest and polyploidy in a p53-mutated human breast cancer cell line. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 376–358. [Google Scholar] [CrossRef]
- Loredana, M.; Ian, O. Tirapazamine: From Bench to Clinical Trials. Cur. Clin. Pharmacol. 2006, 1, 71–79. [Google Scholar] [CrossRef]
- Mattes, W.B.; Hartly, J.A.; Kohn, K.W. Mechanism of DNA strands breakage by piperidine at site of 7N-alkyguanines. Biochim. Biophys. Acta 1986, 868, 71–76. [Google Scholar] [CrossRef]
- Lin, P.S.; Ho, K.C.; Yang, S.J. Tirapazamine (SR4233) interrupts cell cycle progression and induces apoptosis. Cancer Lett. 1996, 105, 249–255. [Google Scholar] [CrossRef]
- Lee, A.E.; Wilson, W.R. Hypoxia-dependent retinal toxicity of bioreductive anticancer prodrugs in mice. Toxicol. Appl. Pharmacol. 2000, 163, 50–59. [Google Scholar] [CrossRef]
- Smitskamp-Wilms, E.; Giaccone, G.; Pinedo, H.M.; Laan, B.F.A.M.; Peters, G.J. DT-diaphorase activity in normal and neoplastic human tissues; an indicator for sensitivity to bioreductive agents? Br. J. Cancer 1995, 72, 917–921. [Google Scholar] [CrossRef]
- Danson, S.; Ward, T.H.; Butler, J.; Ranson, M. DT-diaphorase: a target for new anticancer drugs. Cancer Treat. Rev. 2004, 30, 437–449. [Google Scholar] [CrossRef]
- Workman, P.; Walton, M.I. Enzyme directed bioreductive drug development. In Selective Activation of Drug by Redox Processes; Adams, G.E., Breccia, A., Fielden, E.M., Wardman, P., Eds.; Kluwer Academic/ Plenum Press: New York, NY, USA, 1990; pp. 173–191. [Google Scholar]
- Szmigiero, L.; Kohn, K.W. Mechanism of DNA strand breakage and interstrand cross-linking by diazirinylbenzoquinone (Diaziquone) in isolated nuclei from human cell. Cancer Res. 1984, 44, 4453–4457. [Google Scholar]
- Begleiter, A. Clinical applications of quinone-containing alkylation agents. Front. Biosci. 2000, 5, 153–171. [Google Scholar] [CrossRef]
- Basu, S.; Brown, J.E.; Flannigan, G.M.; Gill, J.H.; Loadman, P.M.; Martin, S.W.; Naylor, B.; Scally, A.J.; Seargent, J.M.; Shah, T.; Puri, R.; Phillips, R.M. Immunohistochemical analysis of NAD(P)H: quinone oxidoreductase and NADPH cytochrome P450 reductase in human superficial bladder tumors: relationship between tumour enzymology and clinical outcome following intravesical mitomycin C therapy. Int. J. Cancer 2004, 109, 703–709. [Google Scholar] [CrossRef]
- Huang, S.T.; Kuo, H.S.; Lin, C.M.; Tsai, H.D.; Peng, Y.C.; and Lin, Y.L. Synthesis and biological evaluation of novel bis-aziridinylnapthoquinone derivatives. Oncol. Res. 2003, 11, 199–204. [Google Scholar]
- Huang, S.T.; Tsai, H.D.; Kuo, H.S.; Yang, Y.P.; Peng, Y.C.; Lin, Y.L. A novel bis-aziridinylnaphthoquinone with anti-solid tumor activity in which induced apoptosis is associated with altered expression of bcl-2 protein. ChemBioChem 2004, 5, 797–803. [Google Scholar] [CrossRef]
- O’Brien, P.J. Molecular mechanisms of quinone cytotoxicity. Chem. Biol. Interact. 1991, 80, 1–41. [Google Scholar] [CrossRef]
- Bulter, J.; Hoey, B.M. Redox cyling drugs and DNA damage. In DNA and Free Radicals; Halliwell, B., Aruoma, O.I., Eds.; Ellis Horwood: London, UK, 1993; pp. 243–265. [Google Scholar]
- Powis, G. Free radical formation by antitumour quinines. Free Radic. Biol. Med. 1989, 6, 63–101. [Google Scholar] [CrossRef]
- Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural Product Hybrides as New Leaders for Drugs Discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028. [Google Scholar] [CrossRef]
- Gauss, W. Uber die umsetzung einiger alkoxy-p-benzochinone mit antylenimin. Chem. Ber. 1958, 91, 2216–2222. [Google Scholar] [CrossRef]
- Eckhardt, G.B.S.J.; Hall, T.C.; Winkler, A. Drug Therapy of Cancer; World Health Organization: Geneva, Switzerland, 1973. [Google Scholar]
- Silva, J.M.; Rao, D.N.; O’Brien, P.J. Modulation of trenimon-induced cytoxicity by DT-disphorase in isolated rat hepatocytes under aerobic versus hypoxic conditions. Cancer Res. 1992, 52, 3015–3021. [Google Scholar]
- Gasparri, F.; Periti, P.; Scarselli, T.; Mazzei, L.; Savino, F.; Branconi, F.; Dancygier, R.G. Perioperative chemotherapy in treatment of carcinoma of uterine cervix. Chemioterapia 1986, 5, 44–52. [Google Scholar]
- Martynov, V.S.; Makarova, A.N.; Berlin, A.Y. Diaminobis(ethylenimino)benzoquinones.II. Reactions of ethyleniminofluoro-1,4-benzoquinones with phenol and derivatives of tyrosin. Akivn. Soedin. Akad. Nauk SSSR. [Chem. Abstr. 1965, 63, 18252b].
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screen. J. Nat. Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Sample Availability: Contact the corresponding author.
© 2009 by the authors. Licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, C.H.; Kuo, H.-S.; Liu, J.-W.; Lin, Y.-L. Synthesis and Antitumor Evaluation of Novel Bis-Triaziquone Derivatives. Molecules 2009, 14, 2306-2316. https://doi.org/10.3390/molecules14072306
Huang CH, Kuo H-S, Liu J-W, Lin Y-L. Synthesis and Antitumor Evaluation of Novel Bis-Triaziquone Derivatives. Molecules. 2009; 14(7):2306-2316. https://doi.org/10.3390/molecules14072306
Chicago/Turabian StyleHuang, Cheng Hua, Hsien-Shou Kuo, Jia-Wen Liu, and Yuh-Ling Lin. 2009. "Synthesis and Antitumor Evaluation of Novel Bis-Triaziquone Derivatives" Molecules 14, no. 7: 2306-2316. https://doi.org/10.3390/molecules14072306
APA StyleHuang, C. H., Kuo, H. -S., Liu, J. -W., & Lin, Y. -L. (2009). Synthesis and Antitumor Evaluation of Novel Bis-Triaziquone Derivatives. Molecules, 14(7), 2306-2316. https://doi.org/10.3390/molecules14072306