2. Modification of the Tetrahydrofuran Moiety
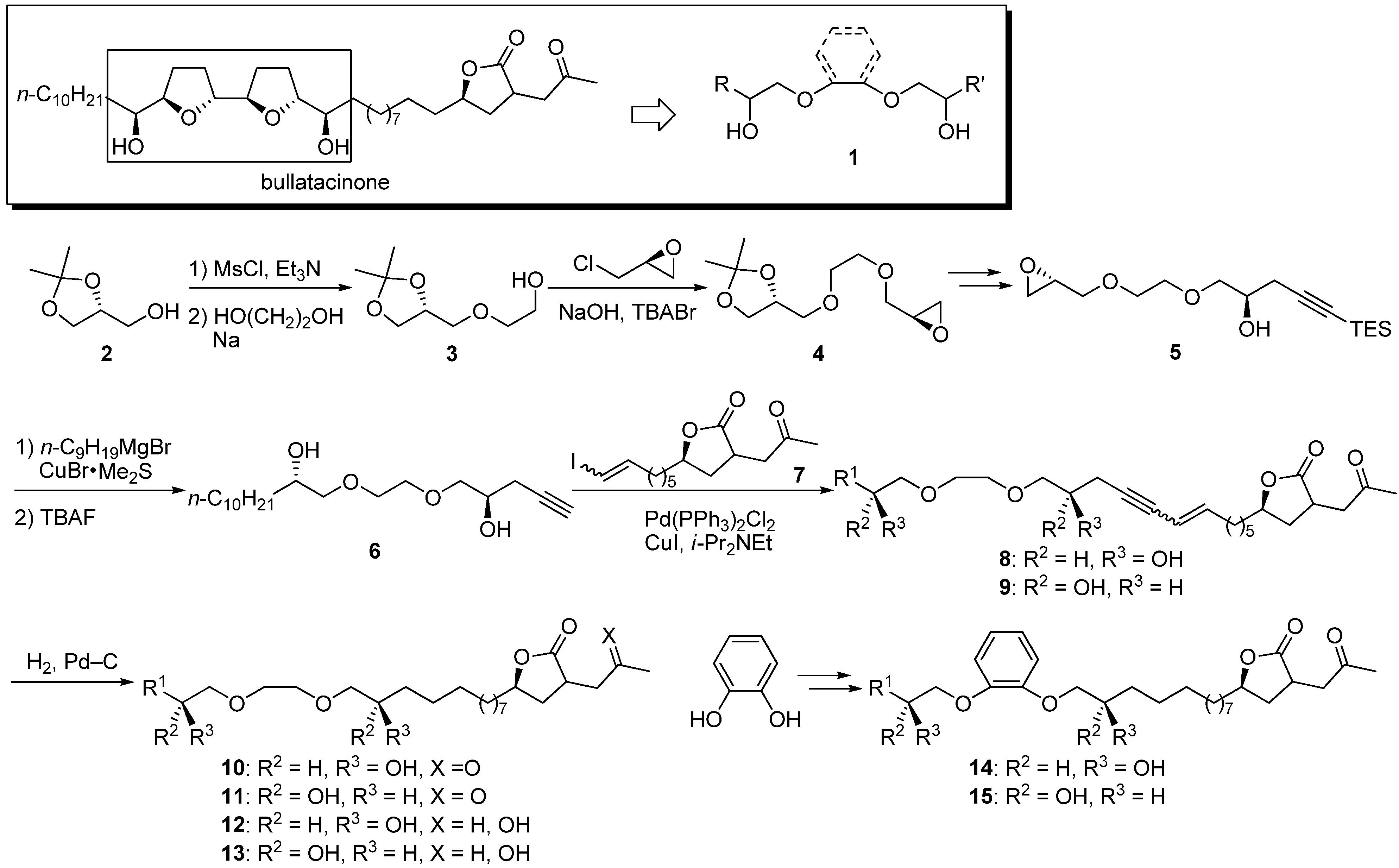
Structural simplification of the tetrahydrofuran moiety, especially the bis-tetrahydrofuran group, is worthwhile because of the limited availability of these complex structures. Grée’s group reported the synthesis and biological activity of a series of acetogenin analogues
1 consisting of ethylene glycol and catechol ethers in place of the bis-tetrahydrofuran core of bullatacinone, which has a ketolactone moiety at the end [
22,
23,
24]. Their analogues were designed to incorporate various lipophilic side chains in place of an
n-alkyl chain. A representative synthetic pathway is given by the preparation of
10a (
Scheme 1). The synthesis of the β-hydroxyl ether core was achieved by condensation of the mesylate of solketal
2, ethylene glycol, and epichlorohydrin. After opening of the epoxide with triethylsilylacetylide, the lipophilic side chain was introduced at the opposite end to give the alkyne
6. Sonogashira coupling of
6 with the γ-lactone fragment
7, followed by hydrogenation, afforded the target analogue
10a. Twenty-three analogues were tested for cytotoxicity against L1210 leukemia cells (
Table 1). Analogues containing catechol were more effective than the series of ethylene glycol derivatives. No significant differences were observed between the various lipophilic side chain substitutions. The simplified acetogenins showed less cytotoxic activity than the natural acetogenins annonacin, bullatacin, and bullatacinone. However, catechol derivatives showed an interesting effect on cell cycle. Natural acetogenins were equally cytotoxic to each phase of the cell cycle, but analogues
14a,
14c–
g, and
15a modified the cell cycle at either phase G1 or G2/M. The target pathway of these analogues may be different from the target of natural acetogenins.
Scheme 1.
Synthesis of ethylene glycol and catechol analogues by Grée’s group.
Scheme 1.
Synthesis of ethylene glycol and catechol analogues by Grée’s group.
Table 1.
IC50 (μM) of synthetic analogues against L1210 cell lines.
Table 1.
IC50 (μM) of synthetic analogues against L1210 cell lines.
| R1 | IC50 | R1 | IC50 |
|---|
| n-C10H21 (8a) | 3.5 | n-C10H21 (9a) | 1 |
| Ph (8b) | 19.8 | Ph (9b) | 21.6 |
| p-MeOC6H4 (8c) | > 10 | p-MeOC6H4 (9c) | 32 |
| p-CF3C6H4 (8d) | 3.3 | – | – |
| 2-Nph (13e) | 2.1 | 2-Nph (9e) | 3 |
| Bu2N (8f) | 3.9 | Bu2N (9f) | 2.5 |
| Oct2N (8g) | 3.7 | – | – |
| N-piperidinyl (8h) | 28.7 | N-piperidinyl (13h) | 23.3 |
| 3-O-cholesteryl (8i) | 2.8 | 3-O-cholesteryl (13i) | 12.2 |
| n-C10H21 (14a) | 1.0 | n-C10H21 (15a) | 7.6 |
| Ph (14b) | 2.0 | – | – |
| p-CF3C6H4 (14d) | 2.2 | doxorubicin | 0.025 |
| 2-Nph (14e) | 0.7 | annonacin | 0.042 |
| Bu2N (14f) | 1.3 | bullatacin | 0.0004 |
| Oct2N (14g) | 2.5 | bullatacinone | 0.016 |
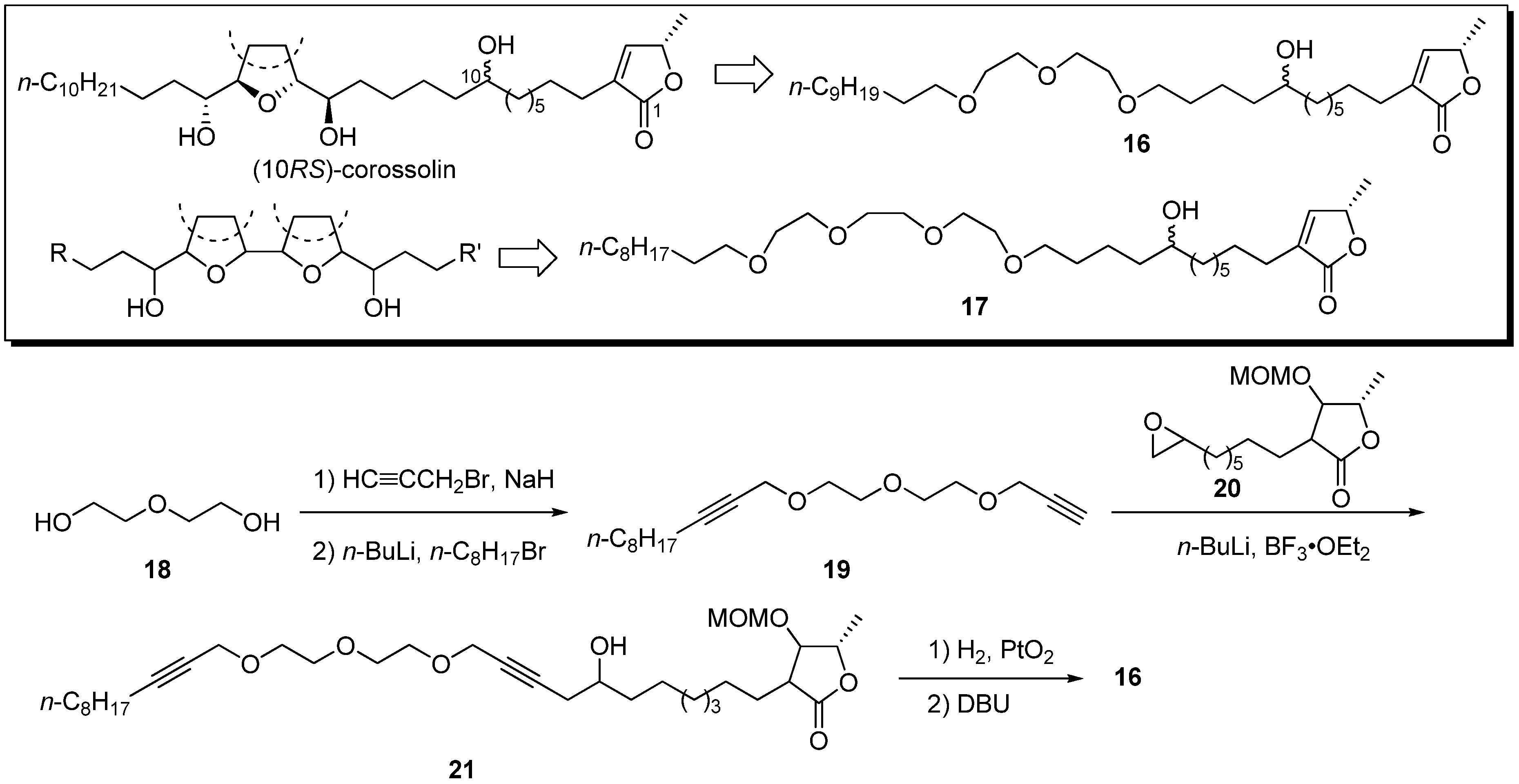
In 2000, Wu’s group reported polyether mimics based on the ionophoric ability [
25,
26,
27] of the THF moiety in acetogenins [
28]. (10
RS)-Corossolin was simplified to the diethylene glycol analogue
16, and the corresponding bis-THF acetogenin, bullatin, was simplified to triethylene glycol
17. A representative synthetic pathway is given by the preparation of
16 (
Scheme 2). Bis-propargylation of diethyleneglycol
18 with propargyl bromide, followed by monoalkylation with
n-octyl bromide, afforded the polyether
19. The reaction of epoxide
20 with the acetylide of
19 yielded the alcohol
21. Hydrogenation of triple bonds, followed by elimination of the MOM-oxy group, afforded analogue
16. Preliminary screening showed that analogues
16 and
17 had moderate activity against HL-60 and K562 (
Table 2).
Scheme 2.
Synthesis of polyether analogues by Wu’s group.
Scheme 2.
Synthesis of polyether analogues by Wu’s group.
Table 2.
In vitro testing against the HL-60 and K562 cell lines.
Table 2.
In vitro testing against the HL-60 and K562 cell lines.
| Conc. (μM) | IG% |
| for HL-60 | for K562 |
| 100 | 10 | 1 | 100 | 10 | 1 |
| 16 | 100 | 50 | 0 | 31 | 18 | 0 |
| 17 | 100 | 65 | 21 | 55 | 25 | 22 |
| corossolone | 68 | 29 | 0 | 53 | 16 | 2 |
| (10RS)-corossolin | 63 | 56 | 5 | 10 | 2 | 0 |
| solamin | 24 | 8 | 0 | 59 | 39 | 29 |
| bullatacin | 73 | 7 | 0 | 53 | 39 | 27 |
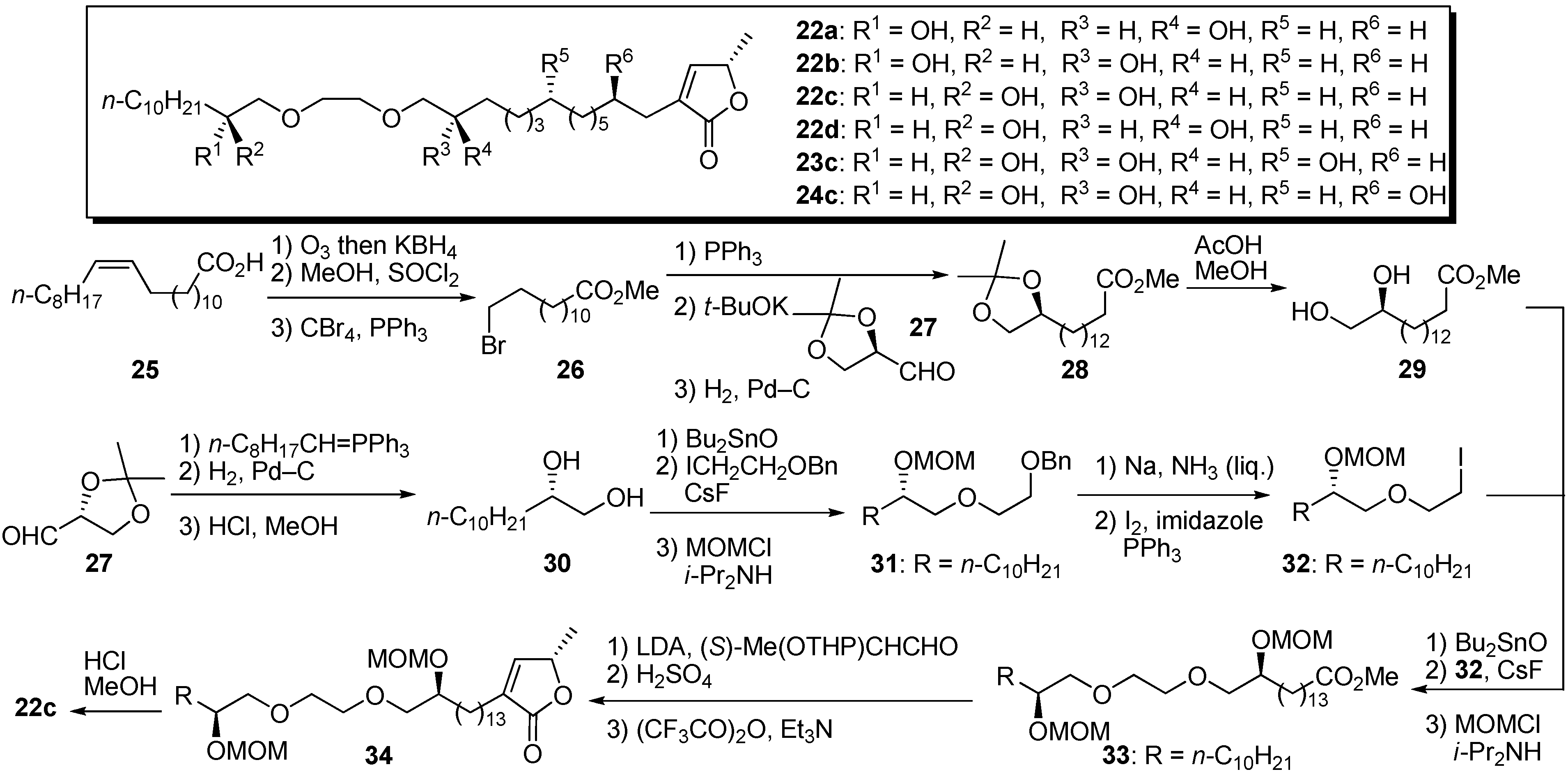
Wu’s group reported the synthesis and biological activity of analogues similar to Gree’s, but Wu’s analogues had an α,β-unsaturated-γ-lactone at the end instead of the ketolactone of Gree’s derivatives [
29,
30]. They completed synthesis of the polyether analogues by a convergent strategy. A representative synthetic pathway is given by the preparation of
22c (
Scheme 3). The synthesis of fragment
29 began with the bromo ester
26 prepared from
cis-erucic acid
25. The Wittig olefination of the phosphonium salt prepared from
26 with (
R)-glyceraldehyde acetonide
27 was followed by hydrogenation and removal of the acetonide protective group, yielding fragment
29. The preparation of the other fragment
32 began with the chain extension of (
R)-glyceraldehyde acetonide
27. The resulting diol
30 was condensed with 2-benzyloxyethyl iodide via a cyclic stannate intermediate. After protection of the secondary alcohol, followed by deprotection of benzyl ether, the resulting primary hydroxyl group was converted to iodide to give
32. The coupling reaction of fragments
29 and
32 was achieved by selective etherification with dibutyltin oxide and cesium fluoride. The γ-lactone moiety was introduced by way of the aldol strategy with
O-THP-(
S)-lactaldehyde. Deprotection of the MOM ether of
34 gave the polyether analogue
22c. The synthesized samples were evaluated by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay to measure cytotoxicity against several human solid tumor cell lines (
Table 3). All four samples showed potent activities against HCT-8 and HT-29 cell lines, whereas they had no cytotoxity against normal human cells. Although the (10
R)-hydroxyl-substituted analogue
23c showed activity similar to
22c, the introduction of the (4
R)-hydroxyl group into
24c raised the potency by a factor of 15 [
31]. It was found that most cell death induced by
22c was due to necrosis, and
23c affected mitochondrial complex I [
32]. A preliminary antitumor assay in mice (Lewis lung cancer) with
22c showed 60% inhibition of tumor compared with the control.
Scheme 3.
Synthesis of polyether analogues by Wu’s group.
Scheme 3.
Synthesis of polyether analogues by Wu’s group.
Table 3.
Cytotoxicity against human solid tumor cell lines.
Table 3.
Cytotoxicity against human solid tumor cell lines.
| Compounds | EC50 [g/mL] |
|---|
| KB | A2780 | HCT-8 | HT-29 |
|---|
| 22a | > 1 | > 1 | 6.6 × 10−2 | 2.72 × 10−1 |
| 22b | > 1 | > 1 | 9.7 × 10−2 | 1.12 |
| 22c | > 1 | > 1 | 3.2 × 10−2 | 1.1 × 10−1 |
| 22d | > 1 | > 1 | 6.5 × 10−2 | 7.83 |
| adriamycin | 2.89 × 10−3 | 1.02 × 10−3 | 4.65 × 10−3 | 9.8 × 10−4 |
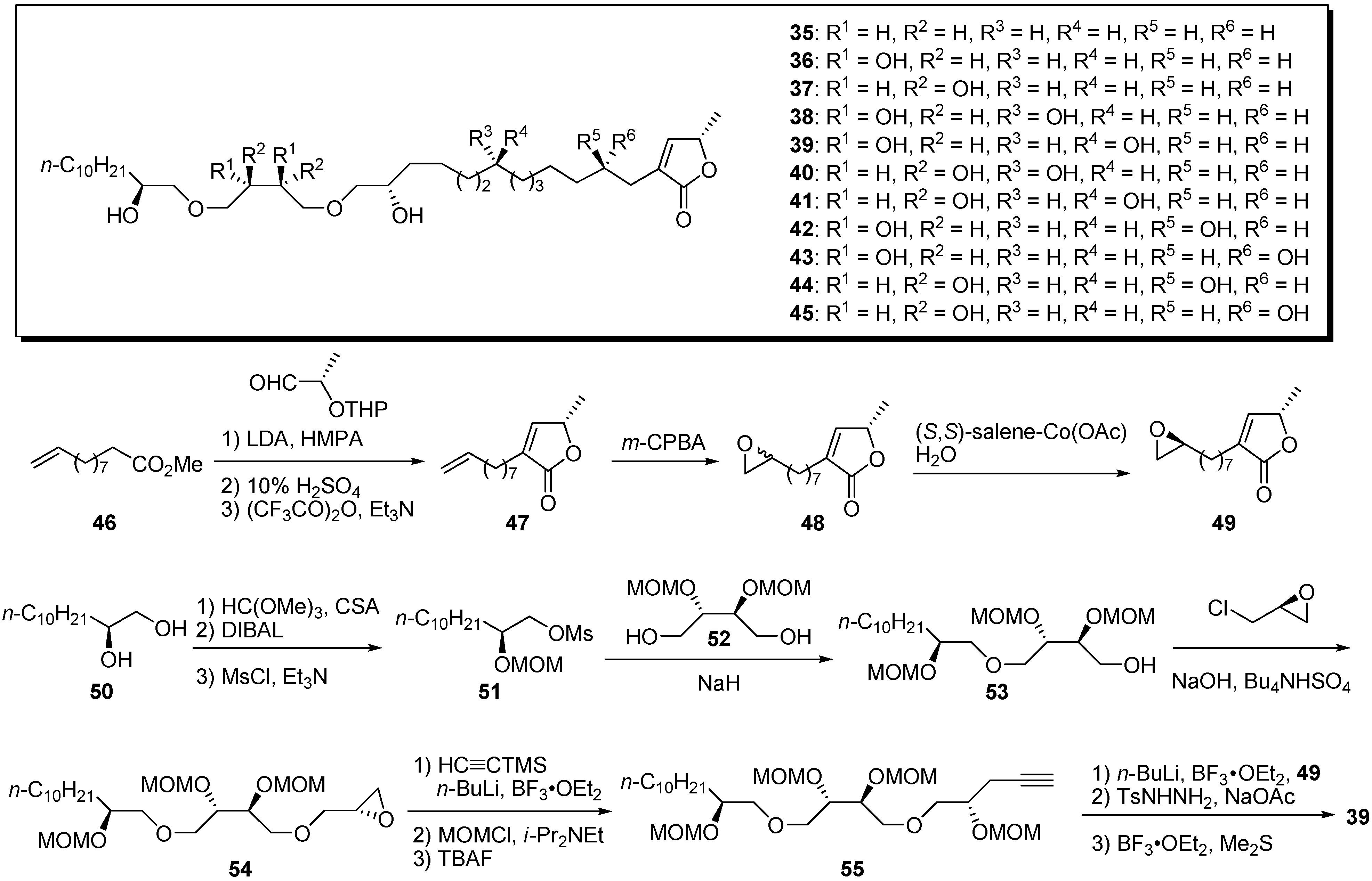
In 2004, Yao and Wu’s group reported preparation of a small library to clarify the structure–activity relationships (SAR) of their polyether analogues [
33]. New analogues that had dihydroxyl groups in the vicinity of the ether bonds were prepared by convergent synthesis. A representative synthetic pathway is given by the preparation of
39 (
Scheme 4). First, ester
46 was transformed to γ-lactone
47 by a three-step sequence involving an aldol reaction with
O-THP-(
S)-lactaldehyde. After epoxidation of the terminal olefin, Jacobsen’s hydrolytic kinetic resolution gave a chiral epoxide
49. The preparation of the polyether fragment
55 began with the condensation of the mesylate
51 and tetraol derivative
52.
O-Alkylation of the alcohol
53 with (
R)-epichlorohydrin afforded the epoxide
54. Opening of the epoxide with trimethylsilylacetylide, followed by protection of the resulting secondary alcohol and deprotection of the TMS group, produced the polyether fragment
55. The coupling reaction of the acetylide prepared from
55 and the epoxide
49, followed by reduction of the triple bond and deprotection of the MOM group, yielded the polyether analogue
39. Nearly all new analogues bearing hydroxyl groups in the vicinity of the ether bonds showed no activity against the Bel-7402 cell line, but exhibited good cytotoxicity against HT-29 and HCT-8 cell lines in the low micromolar range (
Table 4). It is interesting that the introduction of hydroxyl group and their stereochemistry yielded selectivity among the tumor cell lines.
Scheme 4.
Synthesis by Yao and Wu’s group of polyether analogues with dihydroxyl groups in the vicinity of the ether bonds.
Scheme 4.
Synthesis by Yao and Wu’s group of polyether analogues with dihydroxyl groups in the vicinity of the ether bonds.
Table 4.
Cytotoxicity against human solid tumor cell lines.
Table 4.
Cytotoxicity against human solid tumor cell lines.
| Compounds | IC50 [μΜ] |
|---|
| KB | Bel-7402 | HT-29 | HCT-8 |
|---|
| 35 | 7.65 | 1.99 | 0.099 | 0.11 |
| 36 | 4.02 | > 10 | 1.84 | 3.49 |
| 37 | 13.13 | > 10 | 5.72 | 8.58 |
| 38 | 13.81 | > 10 | 7.19 | 5.71 |
| 39 | 23.30 | > 10 | 9.79 | 10.00 |
| 40 | 9.68 | > 10 | 4.56 | 24.46 |
| 41 | 21.30 | > 10 | 7.22 | 6.14 |
| 42 | 6.75 | > 10 | 3.60 | 3.39 |
| 43 | 6.38 | > 10 | 2.36 | 3.51 |
| 44 | 2.00 | > 10 | 1.75 | 2.00 |
| 45 | 2.35 | 4.14 | 1.51 | 3.46 |
| adriamycin | < 0.01 | 0.95 | 0.055 | 0.11 |
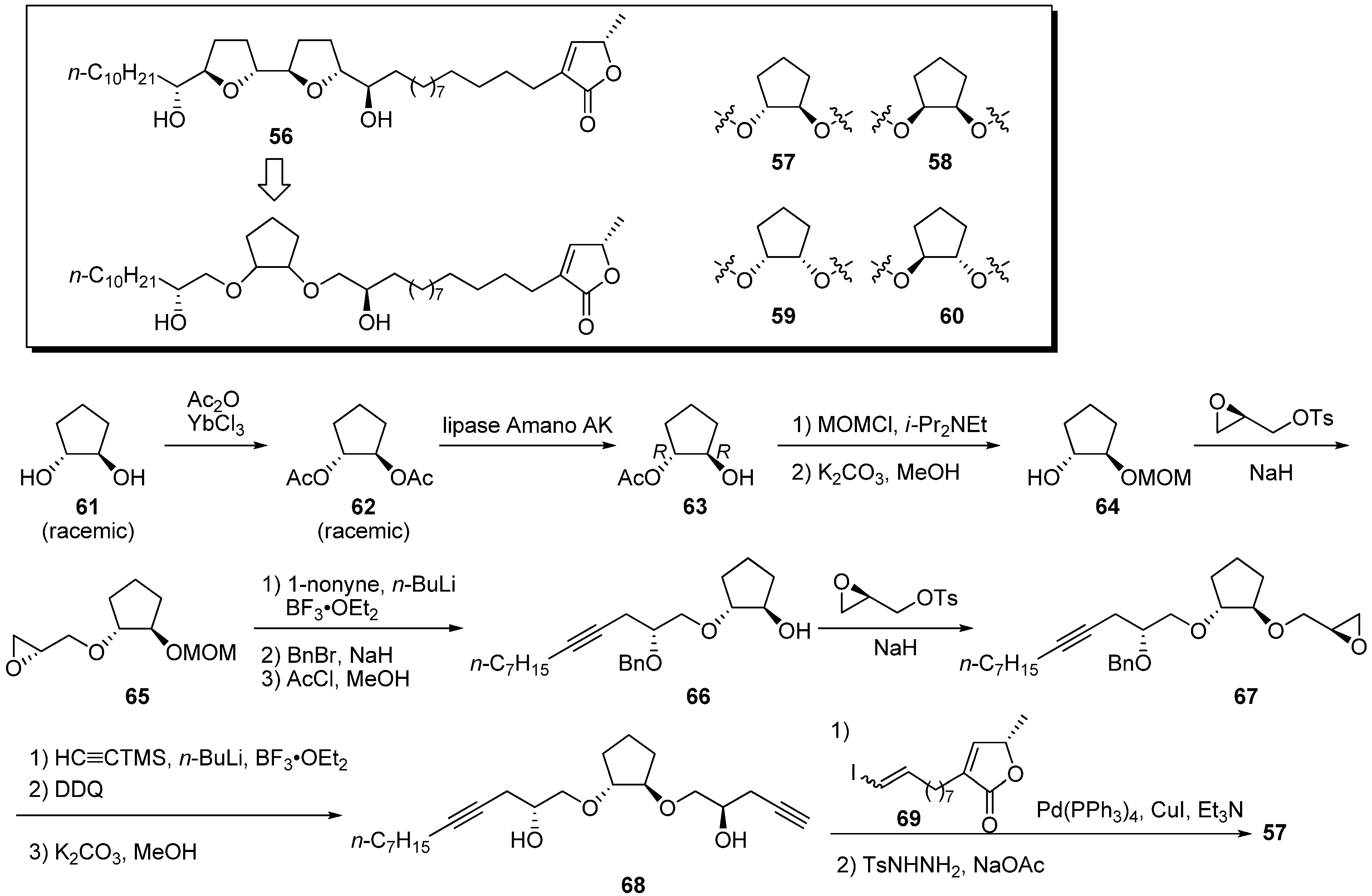
Miyoshi
et al. noted a structural similarity between the hydroxylated bis-THF moiety of natural acetogenins and the hydroxylated 1,2-cyclopentanediol bis-ether motif, especially the relative spatial positions of the four oxygen atoms (
Scheme 5) [
34]. Four diastereomeric 1,2-cyclopentanediol cores were synthesized by optical resolution with lipase. A representative synthetic pathway is given by the preparation of
57. Acetylation of (±)-
trans-1,2-cyclopentandiol
61, followed by hydrolytic optical resolution, gave the chiral monoacetate
63. After protection of the hydroxyl group, treatment with K
2CO
3 in MeOH afforded
64 in the enantiomerically pure form. Introduction of two remaining secondary hydroxyl groups was performed by a coupling reaction with (2
R)-glycidyl tosylate followed by the ring opening of epoxide with acetylide. Sonogashira coupling of alkyne
68 and vinyl iodide
69, followed by selective reduction of the triple bond and enyne, gave the target analogue
57. Inhibitory activities of four mimics against bovine heart mitochondrial complex I were examined (
Table 5). All analogues showed potent inhibition at the nanomolar level, being nearly equipotent with bullatacin, which is one of the most potent inhibitors of complex I. It was also shown that the stereochemistry of 1,2-cyclopentanediol bis-ether cores had a slight effect on inhibitory potency.
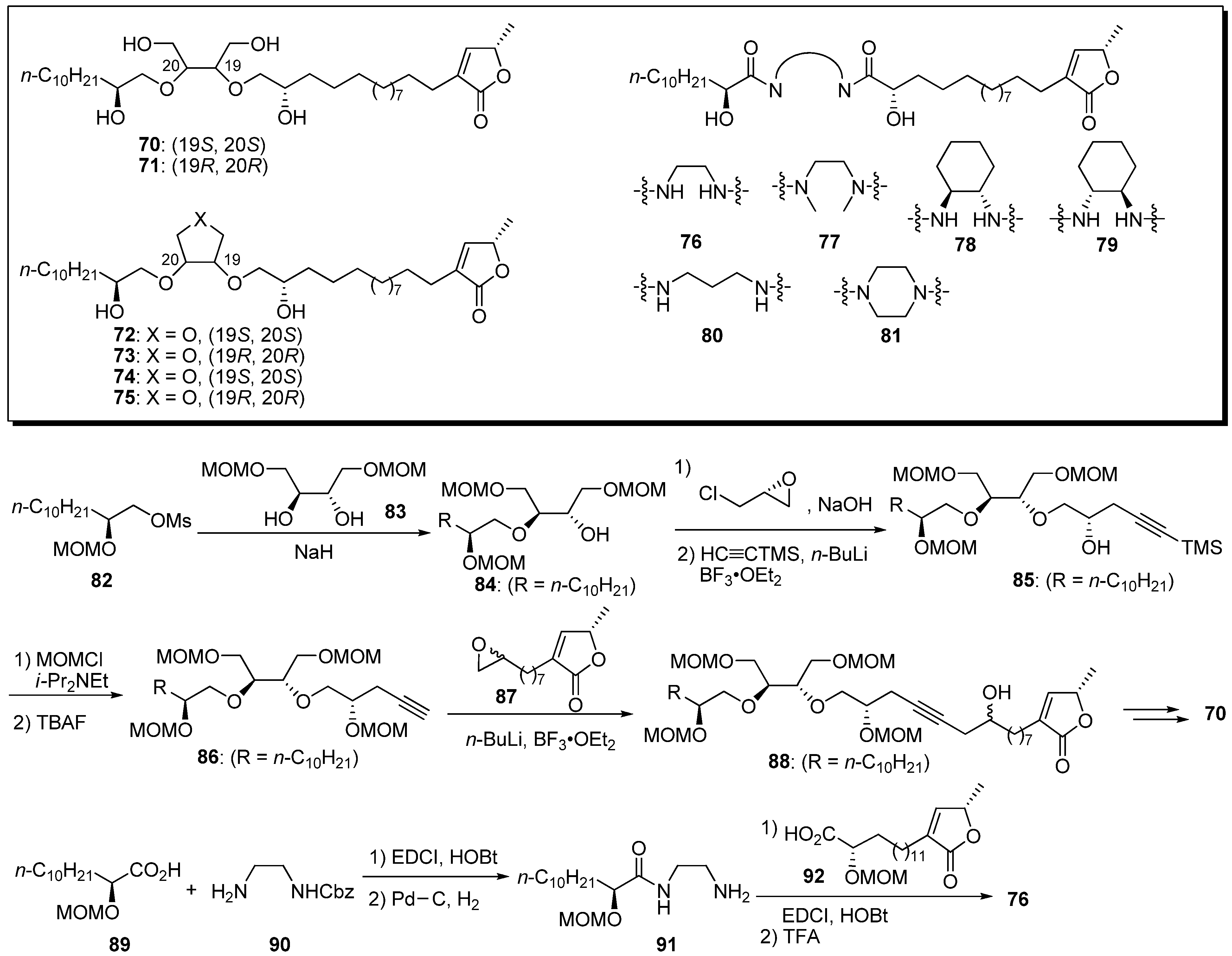
Yao
et al. designed conformationally constrained analogues of the acyclic bis-ether mimics [
35]. The 1,2-disubstitued ethylene glycol, tetrahydrofuran-3,4-diol, tetrahydrothiophene-3,4-diol, and bis-amide moieties were conformationally constrained to alter the ether functionality in the lead compound,
22c. A representative synthetic pathway is given by the preparation of
70 and
76 (
Scheme 6).Synthesis of the analogue
70 with a 1,2-disubstituted ethylene glycol core was started from
O-alkylation of the diol
83 with the mesylate
82, followed by (
R)-epichlorohydrin. The introduction of a γ-lactone moiety was achieved by an epoxide opening reaction with the acetylide of
86. Elimination of the secondary hydroxyl group, followed by selective hydrogenation of the resulting enyne moiety and cleavage of the MOM group, yielded the target analogue
70. The bis-amide analogue
76 was synthesized via sequential coupling of the two carboxylic acids (
89 and
92) with the diamine fragment
90. The inhibitory activities against human breast cancer cell lines, MDA-MB-435 and MDA-MB-468, and non-cancerous human mammary epithelial cells (HMEC) were examined (
Table 6). All analogues, with the exception of
80 and
81, showed low micromolar potencies against MDA-MB-468, whereas they were less active against MDA-MB-435 and displayed satisfactory selectivity for the non-cancerous cell line HMEC. For example, the
N,
N’-dimethyl bis amide derivative
77 (SI = 69), the most potent analogue in this report, showed better selectivity for the inhibition of MDA-MB-468 and HMEC than did its parent
22c (SI = 14). Moreover, compound
77 exhibited 30 times more potency against MDA-MB-468 cell lines than did
22c. These results indicate that the introduction of conformational constraint was useful for the optimization of this class of anticancer agents.
Scheme 5.
Synthesis of 1,2-cyclopentanediol bis-ether analogues by Miyoshi’s group.
Scheme 5.
Synthesis of 1,2-cyclopentanediol bis-ether analogues by Miyoshi’s group.
Table 5.
Summary of the inhibitory potencies (IC50) of the test compounds.a
Table 5.
Summary of the inhibitory potencies (IC50) of the test compounds.a
| Compounds | IC50 (nM) |
| 56 | 0.83 |
| 57 | 1.9 |
| 58 | 1.0 |
| 59 | 1.4 |
| 60 | 0.90 |
| bullatacin | 0.85 |
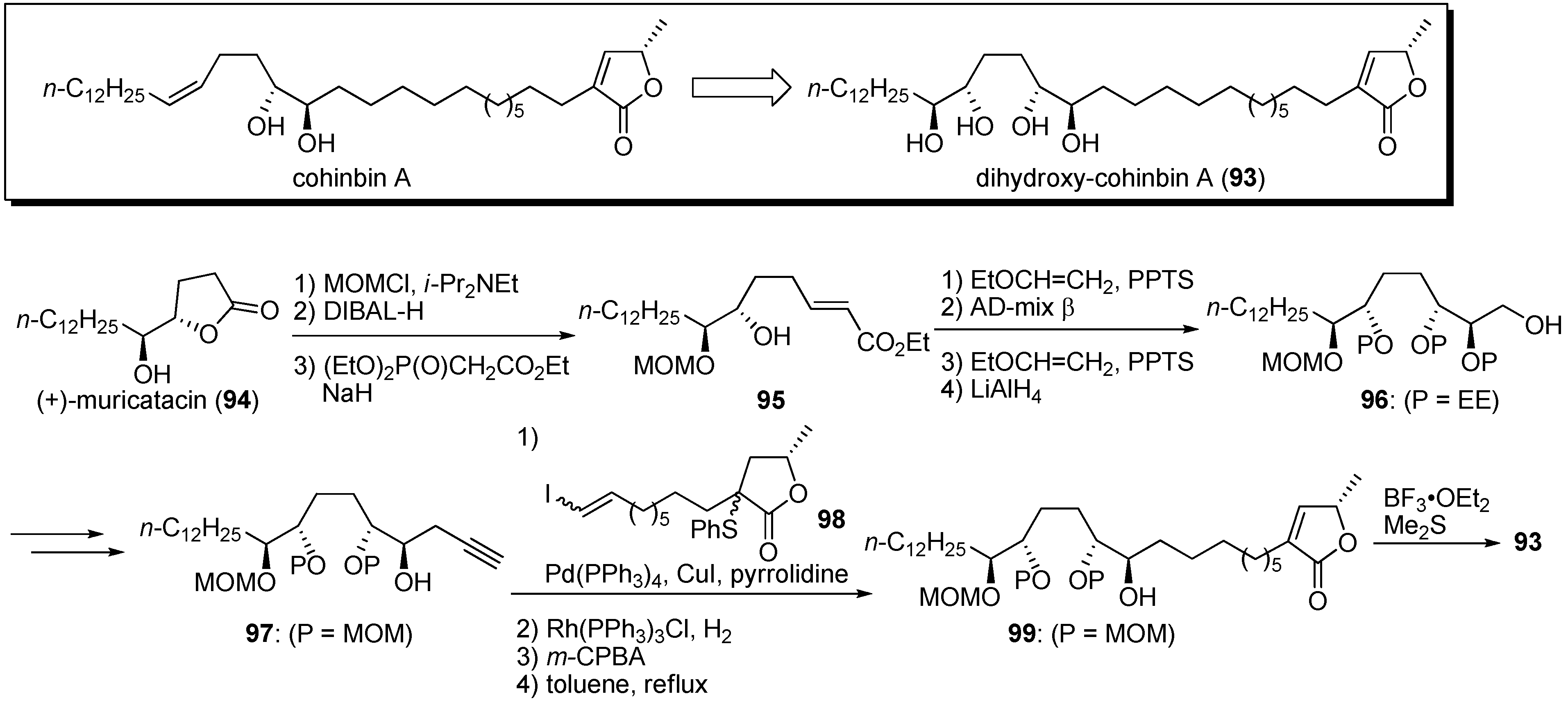
Konno and Miyoshi noted that the THF ring acted as a hydrophilic anchor in the mitochondrial membrane. Dihydroxy-cohinbin A
93 was designed to increase the hydrophilicity of cohinbin A, which belongs to a class of non-THF acetogenins (
Scheme 7) [
36]. The synthesis of dihydroxy-cohinbin A
93 began with (+)-muricatacin
94. The construction of the tetraol moiety was achieved by asymmetric dihydroxylation of the α,β-
E-unsaturated ester with an AD-mix β. Tetraol fragment
97 and γ-lactone fragment
98 were connected by the Sonogashira coupling reaction. After reduction of the enyne moiety, the construction of the α,β-unsaturated-γ-lactone moiety, followed by deprotection of the MOM group, yielded dihydroxy-cohinbin A
93. The inhibitory activities of dihydroxy-cohinbin A
93 and the intermediate
99 against bovine heart mitochondrial complex I were examined (
Table 7). The intermediate
99, which has one free and three MOM-protected hydroxyl groups, lost inhibitory activity. Dihydroxy-cohinbin A
93 showed potent inhibition at the nanomolar level, although its activity was weaker than that of bullatacin. Konno
et al. surmised that bioactivity of dihydro-cohinbin A
93 was diminished by the high degree of flexibility in the tetraol unit compared with the THF unit bearing flanking hydroxyl groups.
Scheme 6.
Synthesis of conformationally constrained polyether analogues by Yao’s group.
Scheme 6.
Synthesis of conformationally constrained polyether analogues by Yao’s group.
Table 6.
Bioactivity screening of newly synthesized analogues.a
Table 6.
Bioactivity screening of newly synthesized analogues.a
| Compounds | IC50[μM] |
|---|
| MDA-MB-435b | MDA-MB-468c | HMECd |
|---|
| 22ce | > 100 | 5.932 | 82.11 |
| 70 | 6.467 | 0.830 | 14.25 |
| 71 | 4.170 | 1.005 | 10.86 |
| 72 | 5.500 | 0.994 | 13.60 |
| 73 | 11.24 | 1.630 | 17.68 |
| 74 | 25.69 | 2.559 | 20.63 |
| 75 | 18.40 | 3.007 | 17.95 |
| 76 | > 100 | 2.953 | > 100 |
| 77 | > 100 | 0.218 | 15.11 |
| 78 | > 100 | 11.81 | > 100 |
| 79 | > 100 | 61.59 | > 100 |
| 80 | > 100 | 2.019 | > 100 |
| 81 | 12.61 | 0.858 | 70.00 |
Scheme 7.
Synthesis of dihydroxy-cohinbin A by Konno and Miyoshi’s group.
Scheme 7.
Synthesis of dihydroxy-cohinbin A by Konno and Miyoshi’s group.
Table 7.
Summary of the inhibitory potencies (IC50) of the test compounds.
Table 7.
Summary of the inhibitory potencies (IC50) of the test compounds.
| Compounds | IC50 (nM) |
|---|
| 93 | 20 |
| 99 | 4100 |
| bullatacin | 0.8 |
3. Modification of the Hydrocarbon Chain
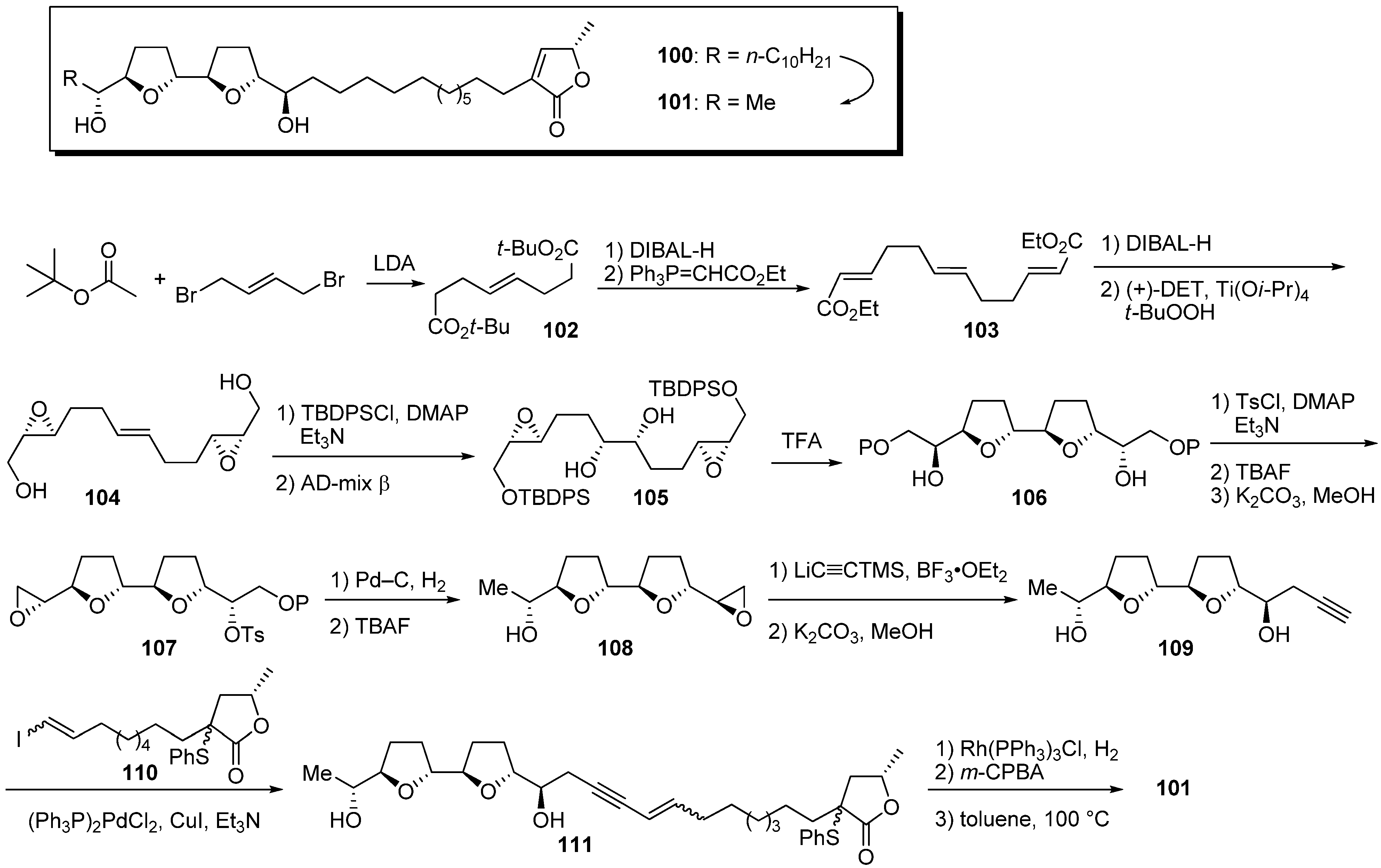
Miyoshi
et al. investigated the role of the hydrophobic alkyl chain that is a feature of natural acetogenins. First, they designed the analogue
101 possessing a methyl group on the left side of the THF ring in place of a long hydrocarbon chain (
Scheme 8) [
37]. The synthesis of the bis-THF fragment began with condensation of
tert-butyl acetate and
trans-1,4-dibromo-2-butene, giving the diester
102. Chain extension of
102, followed by Sharpless asymmetric epoxidation, gave the epoxy alcohol
104. After silylation of the primary alcohol, Sharpless asymmetric dihydroxylation followed by treatment with TFA afforded the bis-THF core
106. The bis-THF core
106 was converted to the epoxide
107 in the following sequential reactions: (1) tosylation of secondary alcohols; (2) mono-desilylation of TBDPS group; (3) treatment with K
2CO
3. Opening of epoxide
107 by hydrogenation with Pd–C, followed by treatment with excess TBAF, yielded alcohol
108. After opening epoxide
108 with trimethylsilylacetylide, followed by desilylation, Pd(0)-mediated coupling of alkyne
109 with vinyl iodide
110 afforded the enyne
111. Hydrogenation of
111 and sequential thermal elimination of the sulfide moiety gave the target analogue
101. Although the IC
50 of
101 (3.1 nM) against bovine heart mitochondrial complex I was weaker than the inhibitory activity of the model compound
100 (0.9 nM), the analogue
101 retained sufficiently potent inhibitory activity (
Table 8). These results indicate that the large hydrophobicity of the left side of the THF ring was not essential for the activity.
Scheme 8.
Synthesis of analogues possessing a methyl group on the left side of the THF ring by Miyoshi’s group.
Scheme 8.
Synthesis of analogues possessing a methyl group on the left side of the THF ring by Miyoshi’s group.
Table 8.
Summary of the inhibitory potencies (IC50) of the test compounds.
Table 8.
Summary of the inhibitory potencies (IC50) of the test compounds.
| Compounds | IC50 (nM) |
|---|
| 100 | 0.9 |
| 101 | 3.1 |
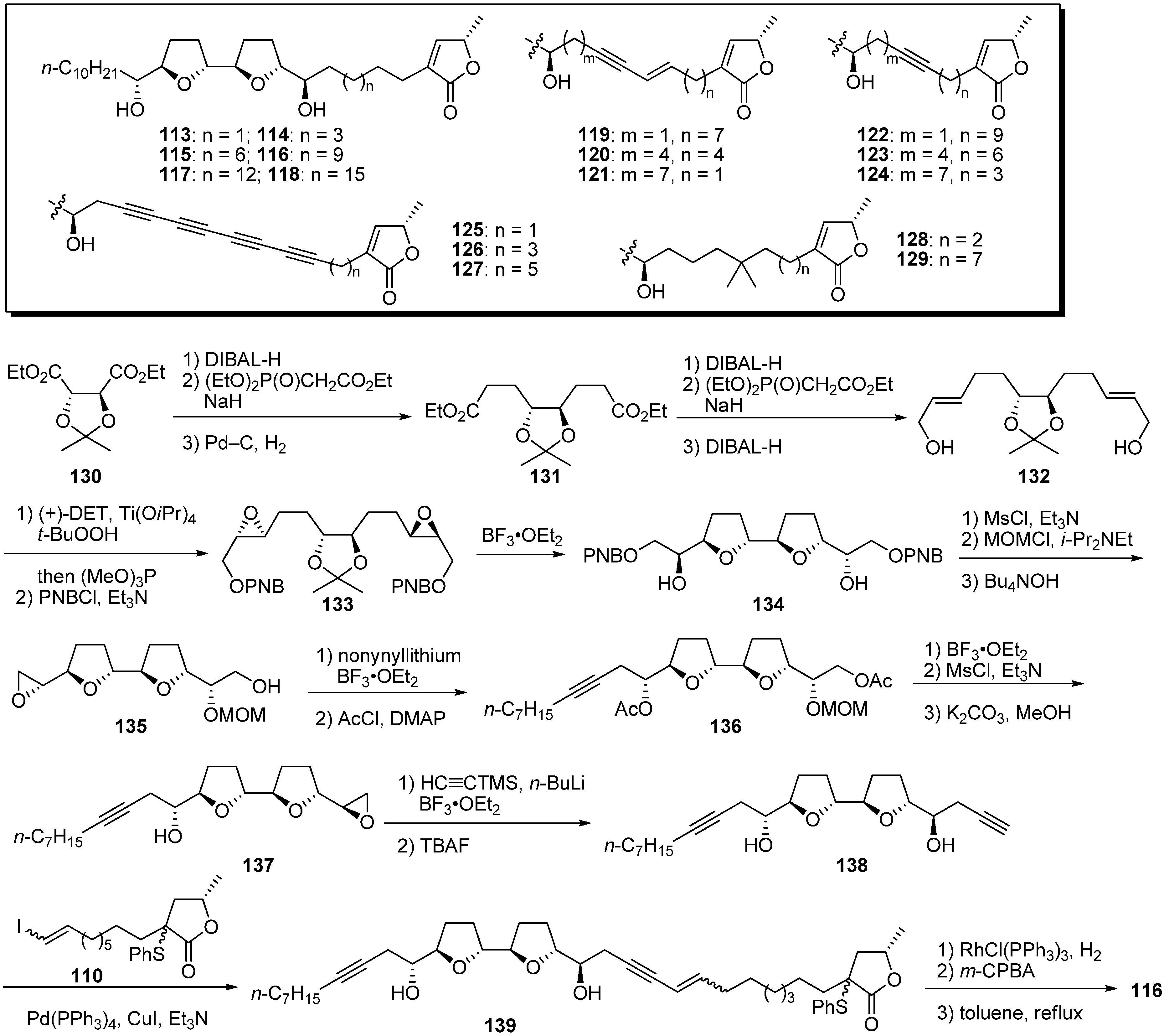
Miyoshi’s group also reported modification of the alkyl spacer linking the THF and γ-lactone rings [
38,
39,
40]. They designed a series of derivatives in which the spacer’s length was varied while other structural factors remained the same, or in which the local flexibility of the spacer was specifically reduced by introducing multiple bond(s) into different regions of the spacer. A representative synthetic pathway is given by the preparation of
116 (
Scheme 9). The synthesis of the THF core
134 started from the diethyl 2,3-
O-isopropylidene-
D-tartrate
130 according to Sasaki
et al. [
41]. After sequential chain extension of
130, Sharpless asymmetric epoxidation of the resulting allyl alcohol
132, followed by treatment with BF
3•OEt
2, gave the bis-THF core
134. Monomesylation of the secondary alcohol of
134, followed by protection of the remaining alcohol and deprotection of the PNB group, yielded the epoxide
135. Treatment of
135 with nonynyllithium, followed by trimethylsilylacetylide, afforded the diyne
138 via
136 and
137. Connection of the two fragments
138 and
110 was achieved via Sonogashira coupling to give
139. Reduction of
139, followed by the formation of the α,β-unsaturated-γ-lactone moiety, gave the target analogue
116. The inhibitory potency of the synthetic analogues was examined (
Table 9). The optimal length of the spacer for inhibition was approximately 13 carbon atoms, which corresponds to the length of spacers in most active natural acetogenins, such as bullatacin. Elongating the spacer beyond 13 carbons reduced inhibitory activity more drastically than did shortening the spacer. Inhibitory potency was not influenced by enhancement of the hydrophobicity (
128 and
129) or local flexibility of the spacer (
119–
124). Surprisingly, tetrayne analogues
125–
127 still exhibited potent inhibition at nanomolar levels, but the double inhibitor titration of complex I activity suggested that the action site of
126 was not identical to that of common acetogenins.
Scheme 9.
Synthesis of analogues possessing modified alkyl spacers linking the THF and γ-lactone rings by Miyoshi’s group.
Scheme 9.
Synthesis of analogues possessing modified alkyl spacers linking the THF and γ-lactone rings by Miyoshi’s group.
Table 9.
Summary of the inhibitory potencies (IC50) of the test compounds.
Table 9.
Summary of the inhibitory potencies (IC50) of the test compounds.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| 113 | 14 | 122 | 1.0 |
| 114 | 1.6 | 123 | 0.83 |
| 115 | 1.2 | 124 | 0.85 |
| 116 | 0.85 | 125 | 6.2 |
| 117 | 13 | 126 | 1.7 |
| 118 | 271 | 127 | 3.0 |
| 119 | 0.92 | 128 | 1.3 |
| 120 | 1.2 | 129 | 1.2 |
| 121 | 1.1 | | |
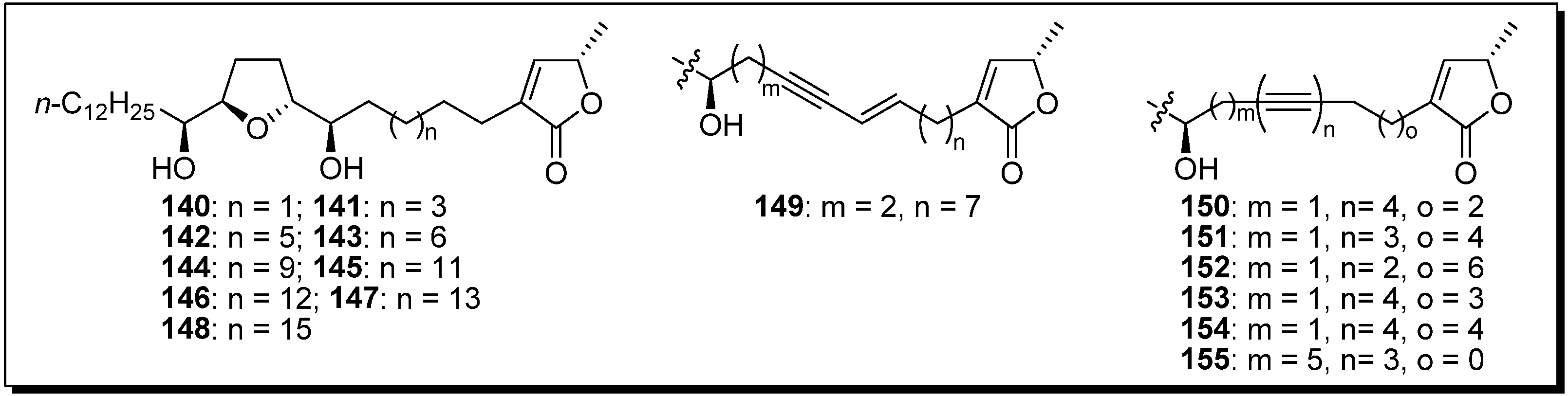
The inhibitory potency of the mono-THF analogue (
144 vs.
150) was drastically reduced, compared with the corresponding bis-THF analogues (
116 vs.
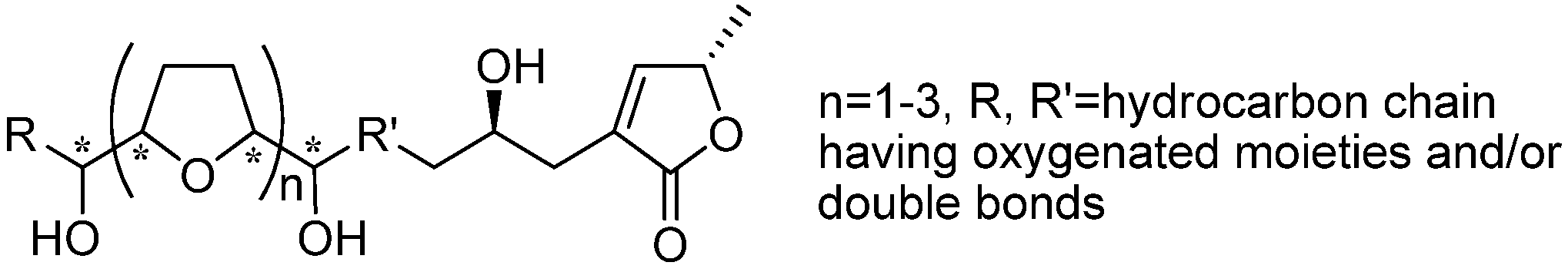
126), by the introduction of a tetrayne structure into the spacer. To clarify the effect of the introduction of tetrayne, Miyoshi
et al. synthesized a new series of tetrayne analogues
150–
155 (
Figure 2). The inhibitory activity of this series showed that the flexibility of the spacer region close to the THF ring was more important than the flexibility of the spacer near the γ-lactone (
Table 10).
Figure 2.
Design of analogues possessing modified alkyl spacers linking the THF and γ-lactone rings, by Miyoshi’s group.
Figure 2.
Design of analogues possessing modified alkyl spacers linking the THF and γ-lactone rings, by Miyoshi’s group.
Table 10.
Summary of the inhibitory potencies (IC50) of the test compounds.
Table 10.
Summary of the inhibitory potencies (IC50) of the test compounds.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| 140 | 131 | 148 | 1050 |
| 141 | 11 | 149 | 5.2 |
| 142 | 10 | 150 | 280 |
| 143 | 10 | 151 | 72 |
| 144 | 2.3 | 152 | 12 |
| 145 | 16 | 153 | 142 |
| 146 | 34 | 154 | 185 |
| 147 | 117 | 155 | 16 |
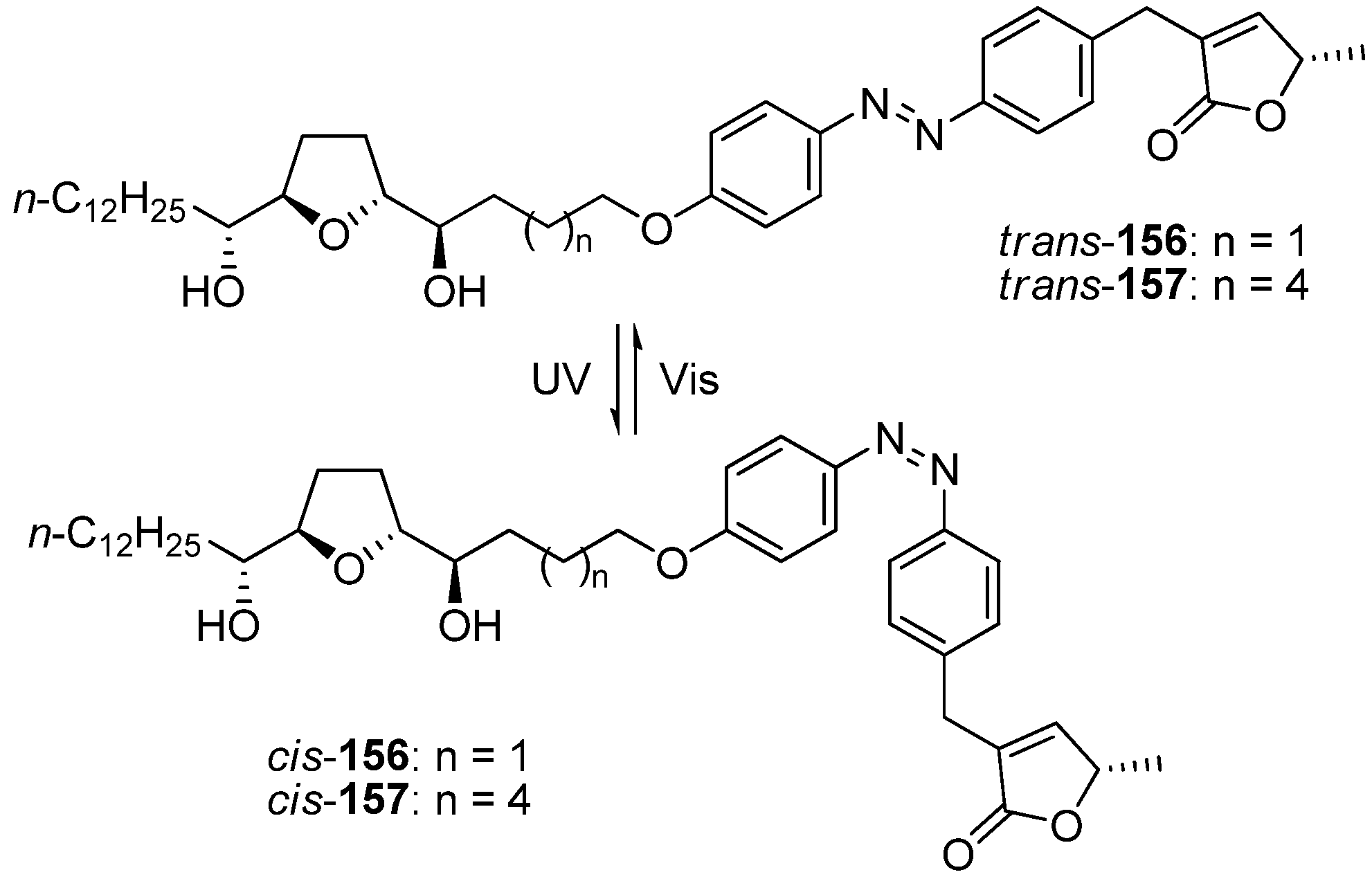
To gain further insight into the function of the spacer, Miyoshi
et al. designed photoresponsive analogues
156–
157 that had an azobenzene moiety in the center of the spacer as a photoresponsive switch (
Scheme 10). The azobenzene unit of
156 reversibly
trans–
cis isomerized by alternating UV-visible irradiation. The NADH oxidase activity of
trans-
156 was weaker than that of
cis-
156.Interestingly, the relative inhibitory effects of the
trans and
cis-
157, which had a longer distance between the THF moiety and the γ-lactone moiety than did
156, were reversed compared with those of
156. As a result of this research, Miyoshi
et al. suggested that acetogenins exhibited potent inhibition of complex I only when the THF moiety and the γ-lactone moiety cooperatively bound to the two putative binding sites. One of the two THF rings in bis-THF acetogenins may have served as a pseudospacer to overcome the significant structural disadvantages that arose from the spacer, whereas mono-THF acetogenins could not efficiently adapt to such structural changes.
Scheme 10.
Photoresponsive analogues possessing an azobenzene moiety in the center of the spacer as a photoresponsive switch.
Scheme 10.
Photoresponsive analogues possessing an azobenzene moiety in the center of the spacer as a photoresponsive switch.
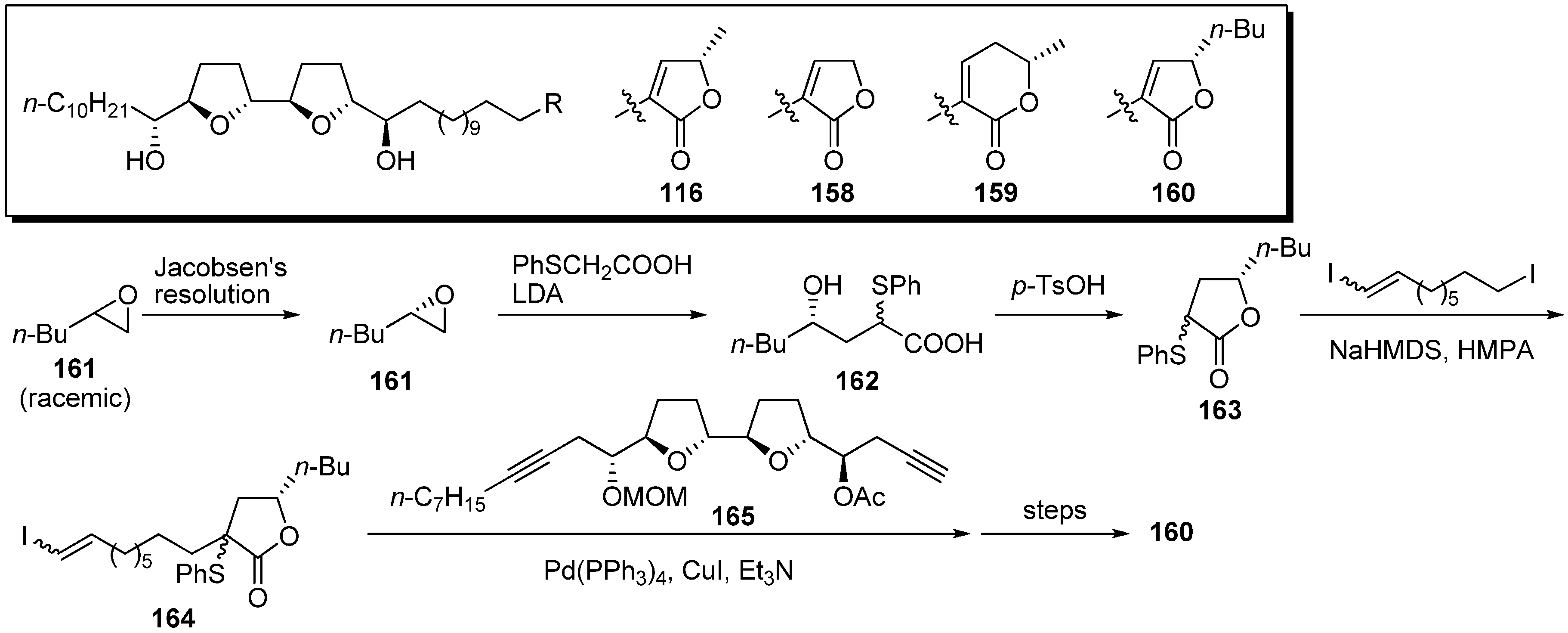
4. Modification of the γ-Lactone Moiety
The γ-lactone moiety in acetogenins was suggested to directly interact with the target site in complex I [
42]. To elucidate the role of the γ-lactone moiety, Miyoshi
et al. synthesized analogues possessing various lactone moieties in place of the α,β-unsaturated-γ-methyl-γ-lactone in natural acetogenins (
Scheme 11) [
43]. A representative synthetic pathway is given by the preparation of
160. Jacobsen’s hydrolytic kinetic resolution of the racemic epoxide
161 gave the chiral epoxide
161. After the epoxide opening of
161 with the dianion prepared from phenylthioacetic acid, lactonization of the resulting seco acid afforded γ-butyl-γ-lactone
163. Sequential assembly of
163, 1,9-diiodononene, and the THF fragment
165, followed by the usual transformation, gave the target analogue
160. The inhibitory activities of synthetic analogues against complex I were examined (
Table 11). The synthetic analogues
158–
160 exhibited inhibition that was as potent as that of the parent compound
116, indicating that the inhibitor binding domain in complex I may be the large cavity-like structure.
Scheme 11.
Synthesis of analogues possessing various lactones by Miyoshi’s group.
Scheme 11.
Synthesis of analogues possessing various lactones by Miyoshi’s group.
Table 11.
Inhibition of mitochondrial complex I.
Table 11.
Inhibition of mitochondrial complex I.
| Compounds | IC50 (nM) |
|---|
| bullatacin | 1.2 |
| 116 | 1.3 |
| 158 | 1.2 |
| 159 | 1.3 |
| 160 | 7.5 |
Acetogenins were proposed to inhibit the terminal electron transfer step of mitochondrial complex I between the Fe-S cluster N2 and the ubiquinone pool [
44,
45,
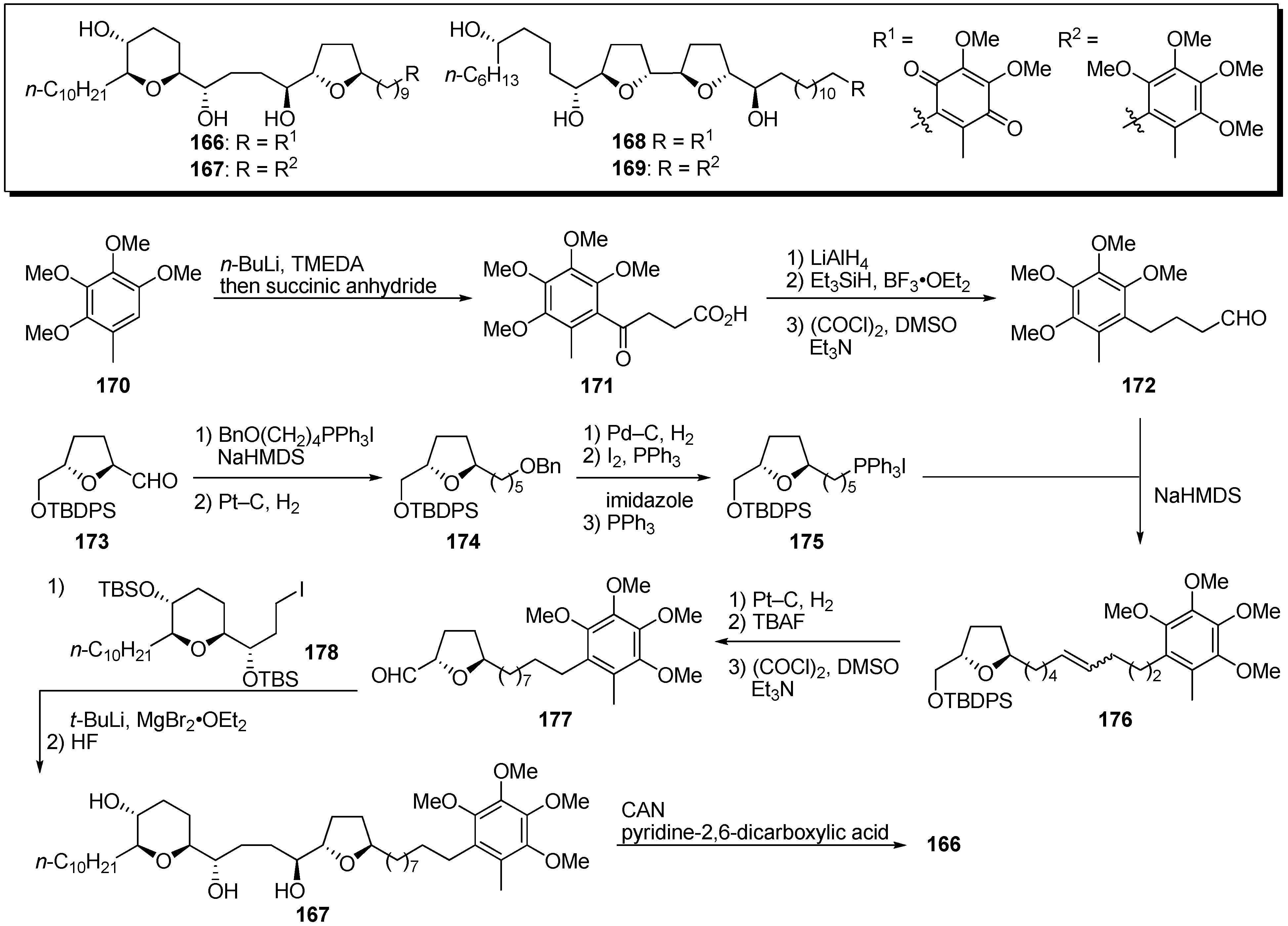
46]. The γ-lactone moiety may bind at the quinone binding site of complex I. To clarify the mode of action of acetogenins, Koert
et al. designed quinone-mucocin
166 and quinone-squamocin D
168 in which the γ-lactone moiety was exchanged for the quinone portion of ubiquinone, the natural substrate of complex I (
Scheme 12) [
47,
48]. A representative synthetic pathway is given by the preparation of
166. The
ortho-lithiation of 2,3,4,5-tetramethoxytoluene
170, followed by treatment with succinic anhydride, gave the carboxylic acid
171. After reduction with LiAlH
4, deoxygenation of the resulting benzylic alcohol, followed by Swern oxidation, afforded aldehyde
172. The THF fragment
175 was prepared from the known aldehyde
173. After chain extension of
173 by the Wittig reaction, followed by hydrogenation, the oxygenated moiety was converted into the phosphonium salt to give
175. Wittig reaction of the aldehyde
172 with the phosphonium salt
175 yielded alkene
176. Introduction of the THP fragment was accomplished by stereoselective coupling of the iodide
178 with the aldehyde
177, prepared from
176. Deprotection of the TBS ethers gave the hydroquinone dimethyl ether
167, which was transformed into the target quinone-mucocin
166 by oxidation with CAN. The hybrid analogues (
166,
168–
169), with the exception of hydroquinone-mucocin
167, were good inhibitors of complex I, and, in particular, quinone-mucocin
166 showed 10 times more potent activity than did mucocin (
Table 12). This result indicated that the γ-lactone moiety in natural acetogenins could be exchanged for the quinone of ubiquinone, although it is unclear that the quinone moiety of the hybrid molecule accepted electrons from complex I.
Scheme 12.
Synthesis of quinone analogues by Koert’s group.
Scheme 12.
Synthesis of quinone analogues by Koert’s group.
Table 12.
Inhibition of mitochondrial complex I.
Table 12.
Inhibition of mitochondrial complex I.
| Compounds | IC50 (nM) |
|---|
| mucocin | 45 |
| 166 | 4.9 |
| 167 | 163 |
| squamocin D | 8.7 |
| 168 | 2.3 |
| 169 | 6.2 |
| rotenone | 1.3 |
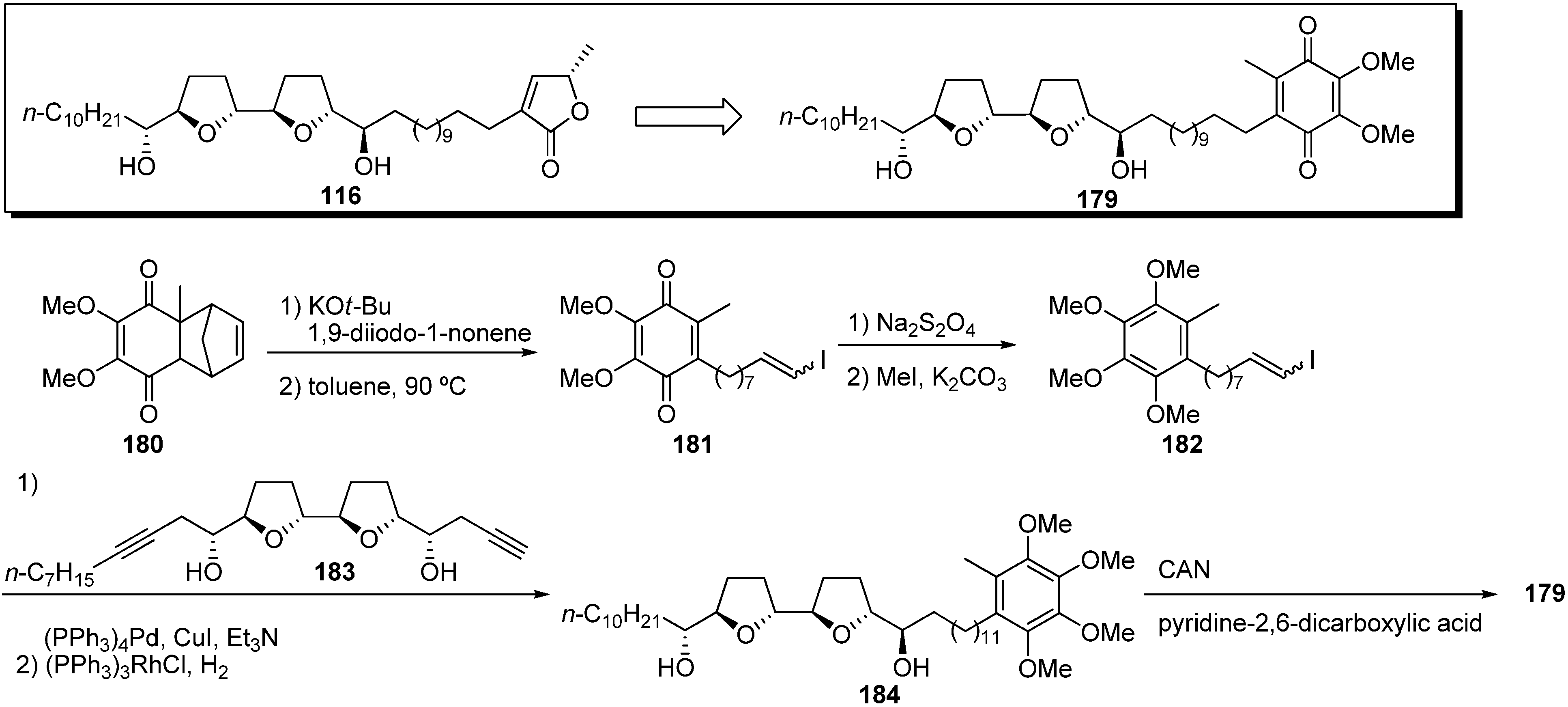
Miyoshi
et al. also synthesized a quinone analogue
179 using the most potent acetogenin
116 synthesized in their laboratory as the mother compound (
Scheme 13) [
49]. Analogue
116 had inhibitory potency equal to bullatacin, the most potent acetogenin. The key fragment
182 was prepared from the known quinone
180 [
50] by alkylation with 1,9-diiodo-1-nonene and a retro-Diels-Alder reaction followed by methylation of the reduced form of quinone
181. Sonogashira coupling of the THF fragment
183 with the vinyl iodide
182, followed by hydrogenation, gave tetramethoxytoluene
184, which was transformed into the quinone analogue
179 by oxidation. The inhibitory activity of
179 was comparable with that of the mother compound
116 or bullatacin (
Table 13). Miyoshi
et al. suggested that the presence of a conjugated carbonyl group may be important for the inhibitory activity of complex I due to the low potency of hydroquinone-dimethyl ether
184. Moreover, the
13C-labeled quinone acetogenins were also synthesized to examine the binding behavior of the quinone group with complex I [
51].
Scheme 13.
Synthesis of quinone analogues by Miyoshi’s group.
Scheme 13.
Synthesis of quinone analogues by Miyoshi’s group.
Table 13.
Inhibitory potencies of test compounds.
Table 13.
Inhibitory potencies of test compounds.
| Compounds | IC50 (nM) |
|---|
| bullatacin | 0.9 |
| 116 | 0.9 |
| 179 | 1.2 |
| 184 | 280 |
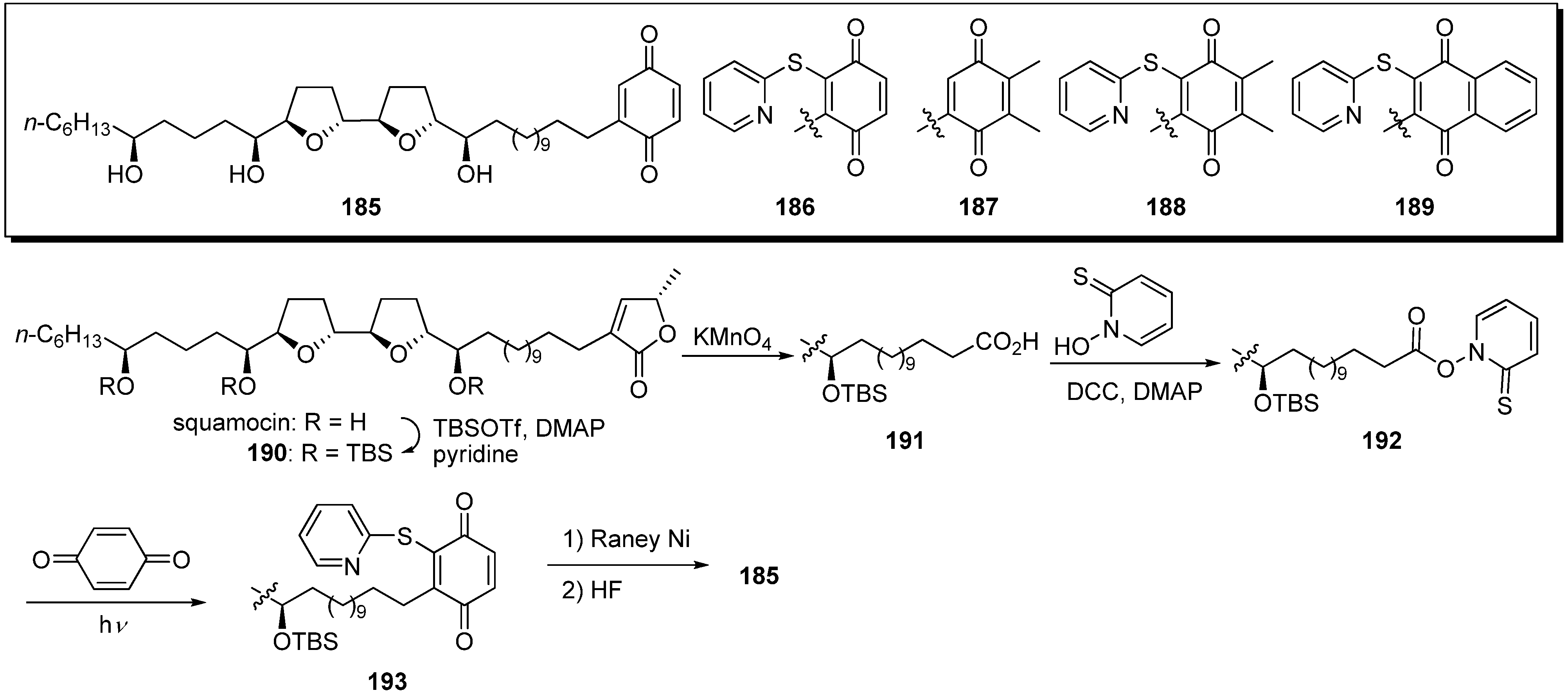
Poupon and Susin
et al. reported the semisynthesis of quinone analogues
185–
189 from natural squamocin (
Scheme 14) [
52]. A representative synthetic pathway is given by the preparation of
185. Treatment of TBS-protected squamocin
190 with KMnO
4 gave the carboxylic acid
191. The condensation of
191 and thiopyridine-
N-oxide with DCC, followed by exposure to light in the presence of the benzoquinone, afforded the thiopyridylquinone derivative
193. After reductive desulfurization of
193 with Raney Ni, the deprotection of the tris-TBS ether yielded the target analogue
185. Screening demonstrated that analogues
185 and
187 possessed a higher pro-apoptotic potential than natural squamocin, whereas the other analogues,
186,
188, and
189, were less effective than squamocin (
Table 14). Moreover, quinone analogues
185 and
187 were potent inhibitors of complex I, although the inhibition activity was weaker than that of squamocin.
Scheme 14.
Semisynthesis of quinone analogues by Poupon’s group.
Scheme 14.
Semisynthesis of quinone analogues by Poupon’s group.
Table 14.
Inhibition of complex I.
Table 14.
Inhibition of complex I.
| Compounds | IC50 (nM) |
|---|
| squamocin | 1.3 |
| rotenone | 30 |
| 185 | 15 |
| 186 | 10 |
| 190 | > 3,000 |
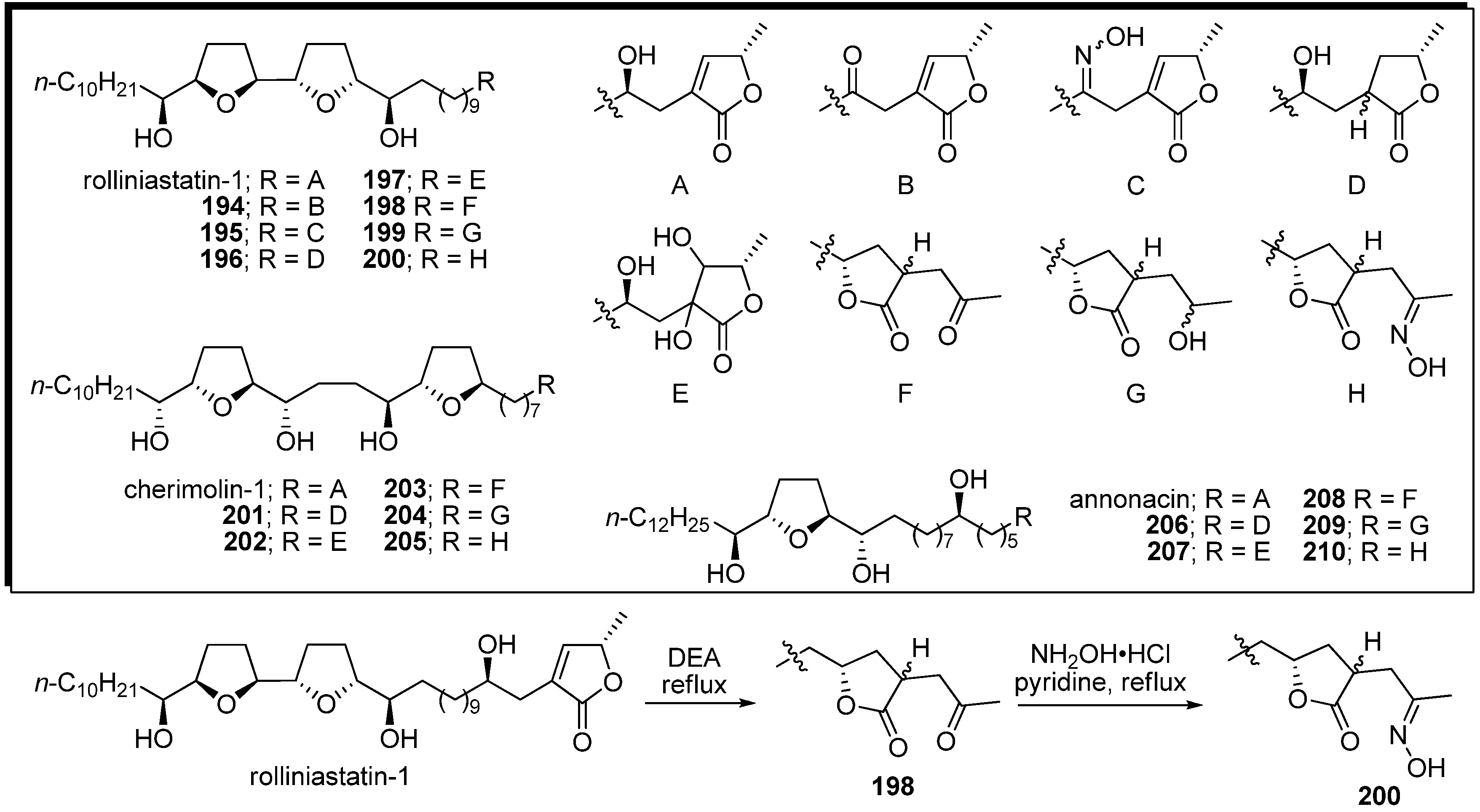
Cortes
et al. reported a series of semisynthetic analogues, modified at the α,β-unsaturated γ-methyl-γ-lactone moiety (
Scheme 15) [
53,
54,
55,
56]. A representative synthetic pathway is given by the preparation of
200. Translactonization of rolliniastatin-1 by alkaline treatment [
57,
58] gave the isoacetogenin analogue
198 as a mixture of 2,4-
cis and 2,4-
trans diastereomers. The carbonyl group of
198 was transformed into the oxime
200 with NH
2OH•HCl in pyridine. The bis-THF acetogenin analogues, with the exception of
202 and
204, indicated more potent inhibitory activity against complex I than the natural compound (
Table 15). Annonacin analogues
206–
210 were tested against some tumor cell lines. Interestingly, the tetrahydroxyl analogue
207, whose inhibitory potency of complex I was the weakest among the annonacin analogues, was the most potent in the cytotoxicity assays.
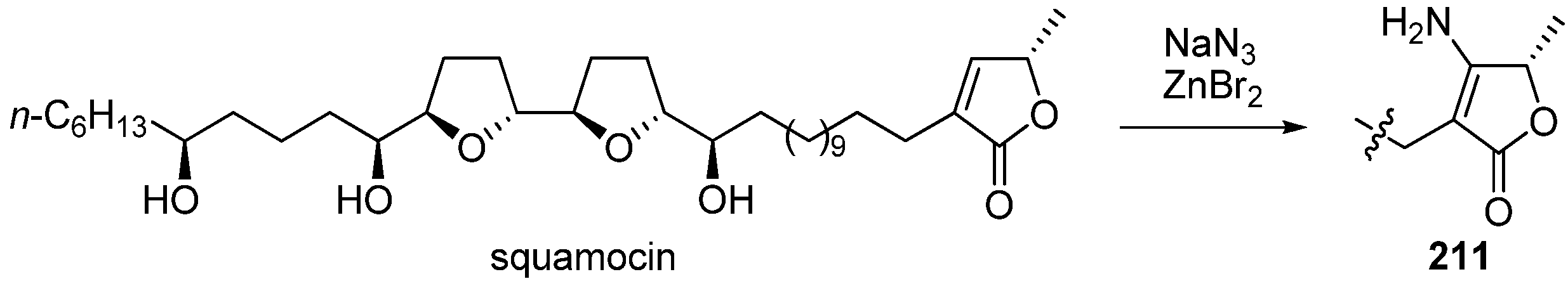
Poupon and Brandt
et al. reported the synthesis and biological evaluation of β-aminosquamocin
211 (
Scheme 16) [
59]. One-step transformation into
211 from squamocin was achieved by treatment with sodium azide and zinc bromide in boiling water. β-Aminosquamocin
211 exhibited more potent cytotoxity against KB3-1 cell lines than did squamocin, despite an inhibitory activity of
211 against complex I that was four times weaker than that of the natural compound (
Table 16). Surprisingly, β-aminosquamocin
211 demonstrated inhibitory activity against complex III at nanomolar levels.
Scheme 15.
Semisynthesis of analogues modified at the α,β-unsaturated γ-methyl-γ-lactone moiety by Cortes’ group.
Scheme 15.
Semisynthesis of analogues modified at the α,β-unsaturated γ-methyl-γ-lactone moiety by Cortes’ group.
Table 15.
Inhibitory potency against complex I.
Table 15.
Inhibitory potency against complex I.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| rolliniastatin-1 | 0.60 | 202 | 8.47 |
| 194 | 0.42 | 203 | 0.83 |
| 195 | 0.25 | 204 | 2.48 |
| 196 | 0.43 | 205 | 0.54 |
| 197 | 0.21 | annonacin | 2.3 |
| 198 | 0.33 | 206 | 3.6 |
| 199 | 0.18 | 207 | 21.8 |
| 200 | 0.23 | 208 | 3.3 |
| cherimolin-1 | 1.84 | 209 | 5.8 |
| 201 | 1.22 | 210 | 1.9 |
Scheme 16.
Synthesis of β-aminosquamocin by Poupon’s group.
Scheme 16.
Synthesis of β-aminosquamocin by Poupon’s group.
Table 16.
Inhibitory activities of 211 and squamocin.
Table 16.
Inhibitory activities of 211 and squamocin.
| Compounds | Cytotoxity (KB3-1)a (M) | Complex I inhibitionb (nM) | Complex III inhibitionc (nM) |
|---|
| squamocin | 1.6 × 10−13 | 2 | inactive |
| 211 | < 10−14 | 8 | 40 |
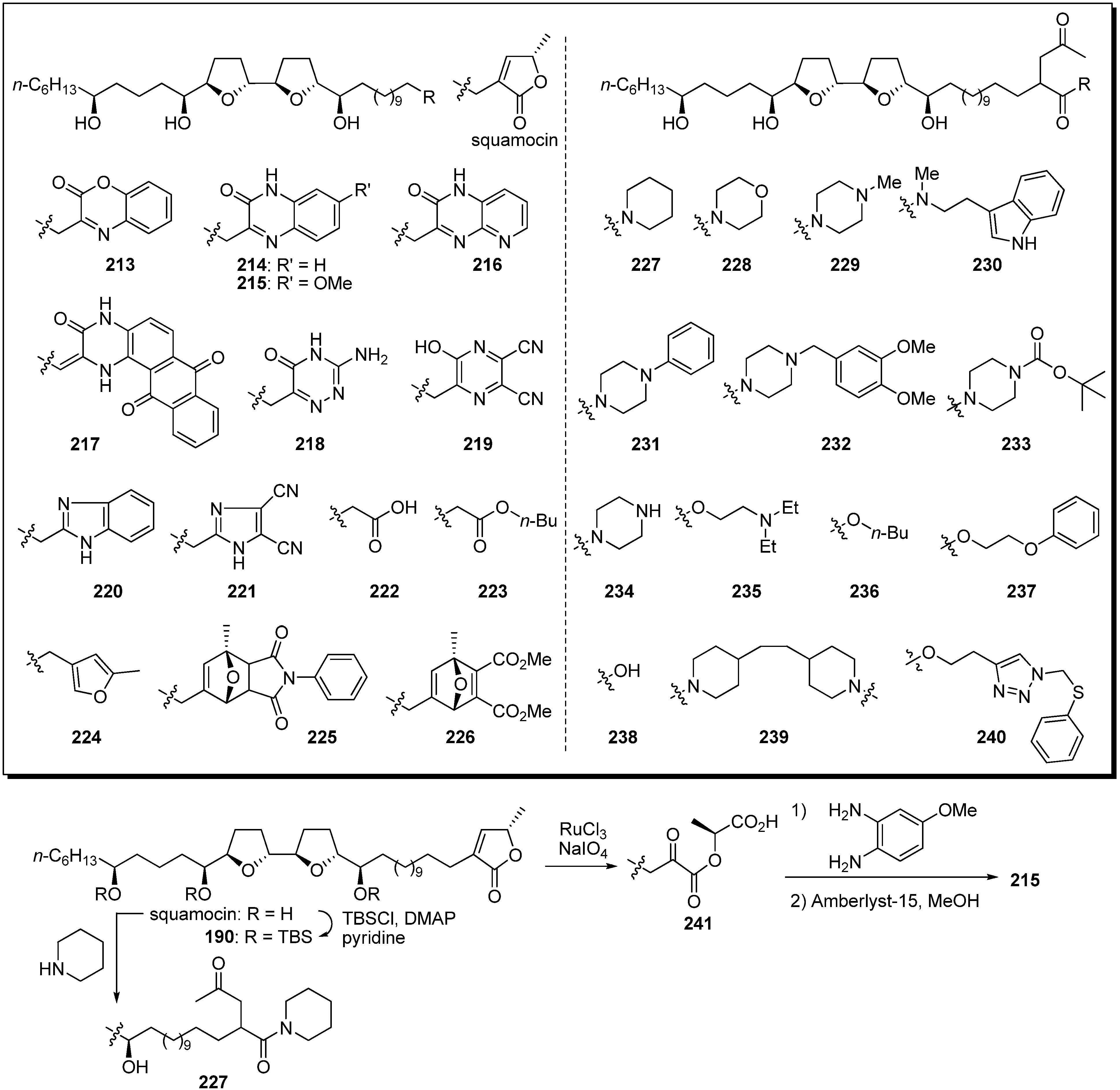
A library of heterocyclic analogues of squamocin was semisynthesized by Lewin
et al., as heterocycles are commonly found as base-structures of potent complex I inhibitors (
Scheme 17) [
60,
61,
62]. A representative synthetic pathway is given by the preparation of
215 and
227. Ruthenium-catalyzed oxidative degradation of terminal γ-lactone in tris-TBS protected squamocin
190 yielded the α-ketoester
241. The condensation of
241 with 4-methoxy-
o-phenylenediamine, followed by deprotection of the TBS ether, afforded the target analogue
215. The γ-ketoamide analogue
227 was synthesized from natural squamocin by treatment with piperidine. The inhibitory activities of heterocyclic analogues
213–
221 against complex I indicated that the γ-lactone moiety in the natural acetogenins could be exchanged for heterocycles (
Table 17). In particular, the benzimidazole analogue
220 had potent inhibitory activity equal to that of squamocin. The α-ketoamide and α-ketoester derivatives, with the exception of
237, showed potent inhibitory activity against complex I at the nanomolar level. Moreover, they had significant cytotoxic activity against KB 3-1 cell lines, although their activities were weaker than that of squamocin.
Scheme 17.
Semisynthesis of squamocin analogues by Lewin’s group.
Scheme 17.
Semisynthesis of squamocin analogues by Lewin’s group.
Table 17.
Biological activities of squamocin analogues.
Table 17.
Biological activities of squamocin analogues.
| | Complex I Inhibition IC50 (nM) | Cytotoxicity (KB 3-1) IC50 (M) | | Complex I Inhibition IC50 (nM) | Cytotoxicity (KB 3-1) IC50 (M) |
| NADH oxidase | NADH:DB oxidoreductase | NADH oxidase | NADH:DB oxidoreductase |
| squamocin | 0.8–0.9 | 1.3 | 1.8 × 10−13 | 227 | 41 | nt | 4.0 × 10−7 |
| 213 | nt | 8.1 | nt | 228 | 22 | nt | 3.6 × 10−7 |
| 214 | 2.0 | 7.9 | nt | 229 | nt | nt | 4.0 × 10−7 |
| 215 | nt | 10 | nt | 230 | 52 | nt | 3.9 × 10−7 |
| 216 | nt | 17 | nt | 231 | nt | nt | 1.5 × 10−8 |
| 217 | 13 | nt | nt | 232 | 74 | 154 | 1.2 × 10−8 |
| 218 | 19 | nt | nt | 233 | nt | nt | 4.1 × 10−8 |
| 219 | 122 | nt | nt | 234 | nt | nt | 5 × 10−7 |
| 220 | 0.9 | 3.3 | nt | 235 | 13 | nt | 7.5 × 10−10 |
| 221 | 14 | 41 | nt | 236 | 17 | nt | 4.6 × 10−10 |
| 222 | 6.2 | 38 | 2.8 × 10−7 | 237 | > 3000 | nt | 8.1 × 10−8 |
| 223 | 2.3 | 9.2 | 6.8 × 10−8 | 238 | 12 | nt | 1.3 × 10−9 |
| 224 | 2.4 | nt | 3.2 × 10−8 | 239 | 26 | nt | 2.7 × 10−8 |
| 225 | 30 | nt | 3.5 × 10−7 | 240 | 12 | nt | 5.1 × 10−9 |
| 226 | 23 | nt | 3.0 × 10−8 | | | | |
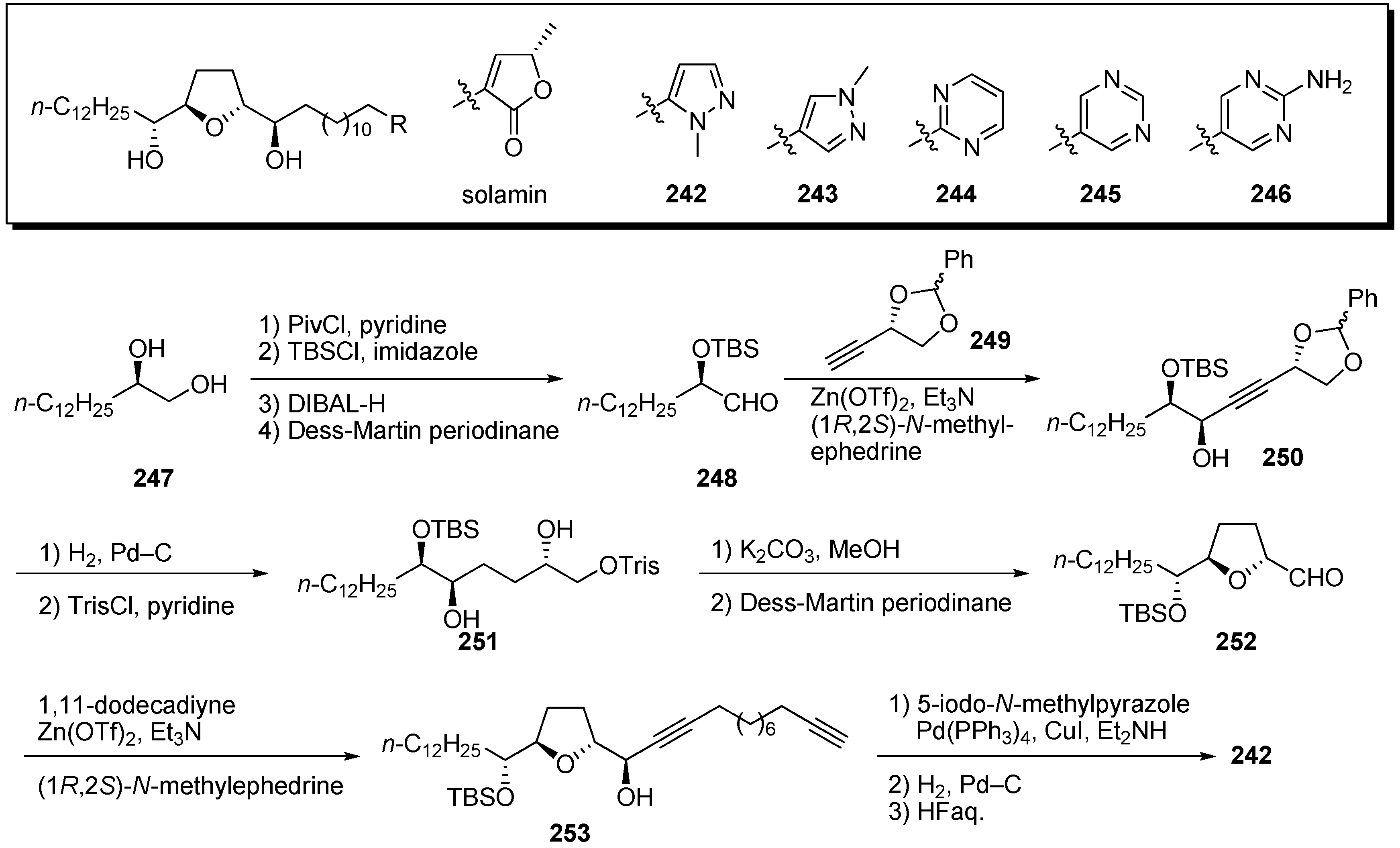
Kojima and Tanaka
et al. designed and synthesized heterocyclic analogues of solamin, a simple mono-THF acetogenin (
Scheme 18) [
63]. A representative synthetic pathway is given by the preparation of
242.
Scheme 18.
Synthesis of heterocyclic solamin analogues by Tanaka’s group.
Scheme 18.
Synthesis of heterocyclic solamin analogues by Tanaka’s group.
1,2-Diol
247 was converted to the α-TBS-oxyaldehyde
248 via the following sequential reactions: (1) Esterification of the primary alcohol; (2) silylation of the remaining secondary alcohol; (3) deprotection of pivaloyl ester; 4) oxidation of the resulting primary alcohol. The introduction of a chiral C4-unit
249 was achieved by asymmetric alkynylation with a Zn(OTf)
2/Et
3N system to give
250 [
64]. Reduction of the triple bond and deprotection of the benzylideneacetal, followed by selective sulfonylation of the primary alcohol, gave sulfonate
251. Treatment of
251 with K
2CO
3, followed by oxidation of the resulting primary alcohol, afforded the THF ring
252. Alkynylation of
252 with 1,11-dodecadiyne proceeded smoothly to give the propargy alcohol
253. After introduction of the aromatic ring by the Sonogashira reaction, hydrogenation of diyne moiety, followed by deprotection, yielded the target analogue
242. Synthetic analogues
242–
246 were tested for
in vitro antiproliferative activity against a panel of 39 human cancer cell lines [
65]. Selected GI
50 (concentration for 50% inhibition of cell growth relative to control) values are summarized in
Table 18. The
N-methylpyrazole derivative
242 displayed strong cytotoxicity against NCI-H23 with potencies that were 80 times higher than those of solamin. These results indicate that the γ-lactone moiety could be exchanged for a heterocycle and that antitumor agents more effective than the natural acetogenins could be produced.
Table 18.
Selected GI50 (M) values of heterocyclic analogues against human cancer cell lines.
Table 18.
Selected GI50 (M) values of heterocyclic analogues against human cancer cell lines.
| Compounds | MCF-7a | SF-295b | HCT-116c | NCI-H23d | OVCAR-4e | MKN7f | PC-3g |
|---|
| solamin | > 10−4 | 4.0 × 10−5 | > 10−4 | 7.3 × 10−5 | > 10−4 | 1.3 × 10−5 | > 10−4 |
| 242 | 2.7 × 10−5 | 2.4 × 10−5 | 9.2 × 10−6 | 9.1 × 10−7 | 3.7 × 10−5 | 6.2 × 10−6 | > 10−4 |
| 243 | > 10−4 | 2.4 × 10−5 | 7.0 × 10−5 | 1.3 × 10−5 | > 10−4 | 5.4 × 10−6 | > 10−4 |
| 244 | 7.9 × 10−5 | > 10−4 | > 10−4 | 2.8 × 10−5 | 3.8 × 10−5 | 2.7 × 10−5 | 6.8 × 10−5 |
| 245 | 3.4 × 10−5 | 2.5 × 10−5 | 2.8 × 10−5 | 1.1 × 10−5 | 2.2 × 10−5 | 6.1 × 10−6 | 3.5 × 10−5 |
| 246 | > 10−4 | 5.6 × 10−5 | > 10−4 | 3.6 × 10−5 | > 10−4 | 9.4 × 10−6 | > 10−4 |
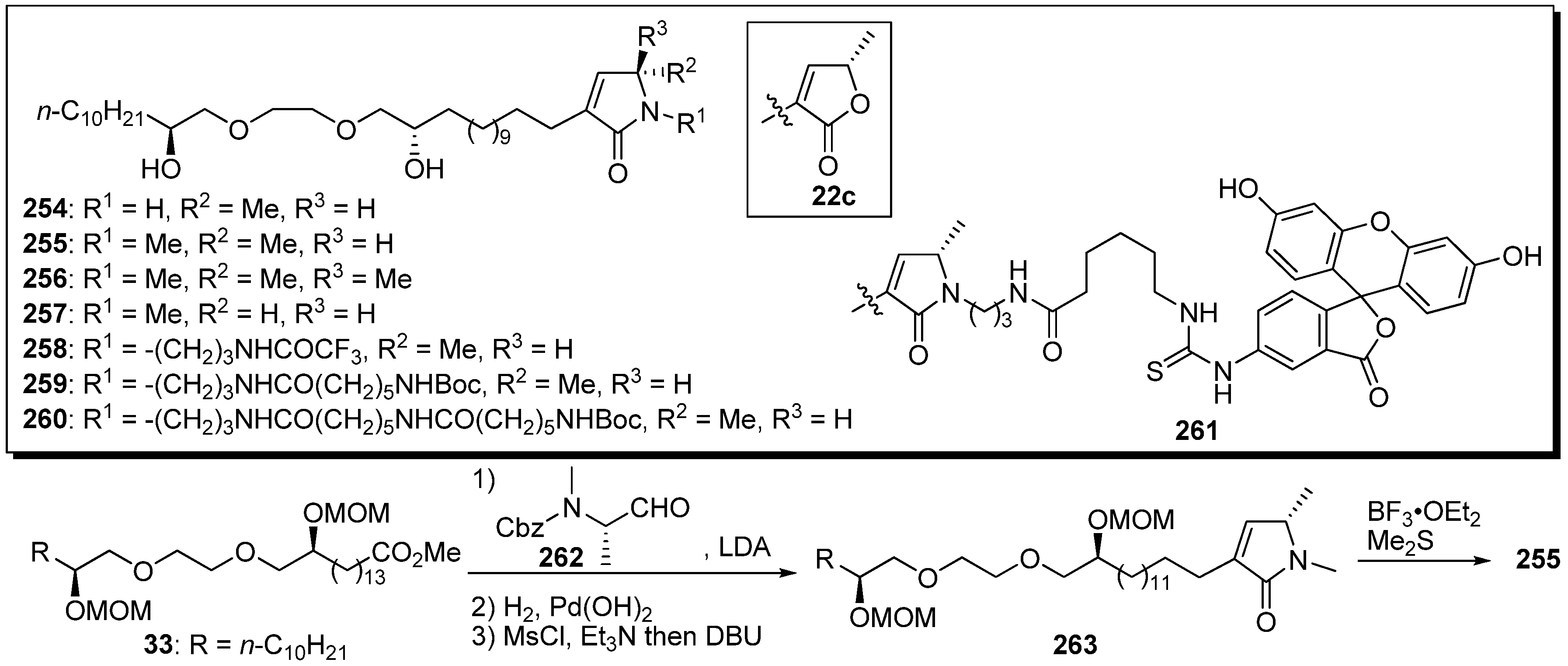
Yao
et al. reported the synthesis of new analogues bearing a terminal lactam moiety instead of the γ-lactone in their original polyether mimics
22c (
Scheme 19) [
66]. A representative synthetic pathway is given by the preparation of
255. Aldol reaction of the enolate from the ester
33 with amino aldehyde
262, removal of the Cbz group, and
in situ cyclization with β-elimination gave the lactam
263. Deprotection of the MOM ethers afforded the target compound
255.
N-Methyl substituted lactam derivatives
255–
257 showed potent cytotoxicity and good selectivity between human cells and tumor cells (
Table 19). Unfortunately, analogues
258–
260, with different length of the linkers at the nitrogen atom of the terminal lactam, showed dramatically decreased cytotoxic activity compared with
255, and no activity was measured in the fluorescent probe
261.
Scheme 19.
Synthesis of lactam-containing analogues by Yao’s group.
Scheme 19.
Synthesis of lactam-containing analogues by Yao’s group.
Table 19.
Cytotoxicity screening of lactam-containing analogues.
Table 19.
Cytotoxicity screening of lactam-containing analogues.
| Compounds | IC50 (µM) |
|---|
| | Chang | B16 | BEL-7404 | SK-Hepl |
| 22c | NAa | 0.035 | 0.041 | 0.065 |
| 254 | NA | 0.87 | 2.20 | NA |
| 255 | NA | 0.013 | 0.234 | 0.589 |
| 256 | NA | 0.478 | 0.845 | 0.583 |
| 257 | NA | 0.168 | 0.168 | 0.104 |
| 258 | NA | 0.995 | 1.35 | NA |
| 259 | NA | NA | 3.20 | NA |
| 260 | NA | NA | 2.50 | NA |
| 261 | NA | NA | NA | NA |
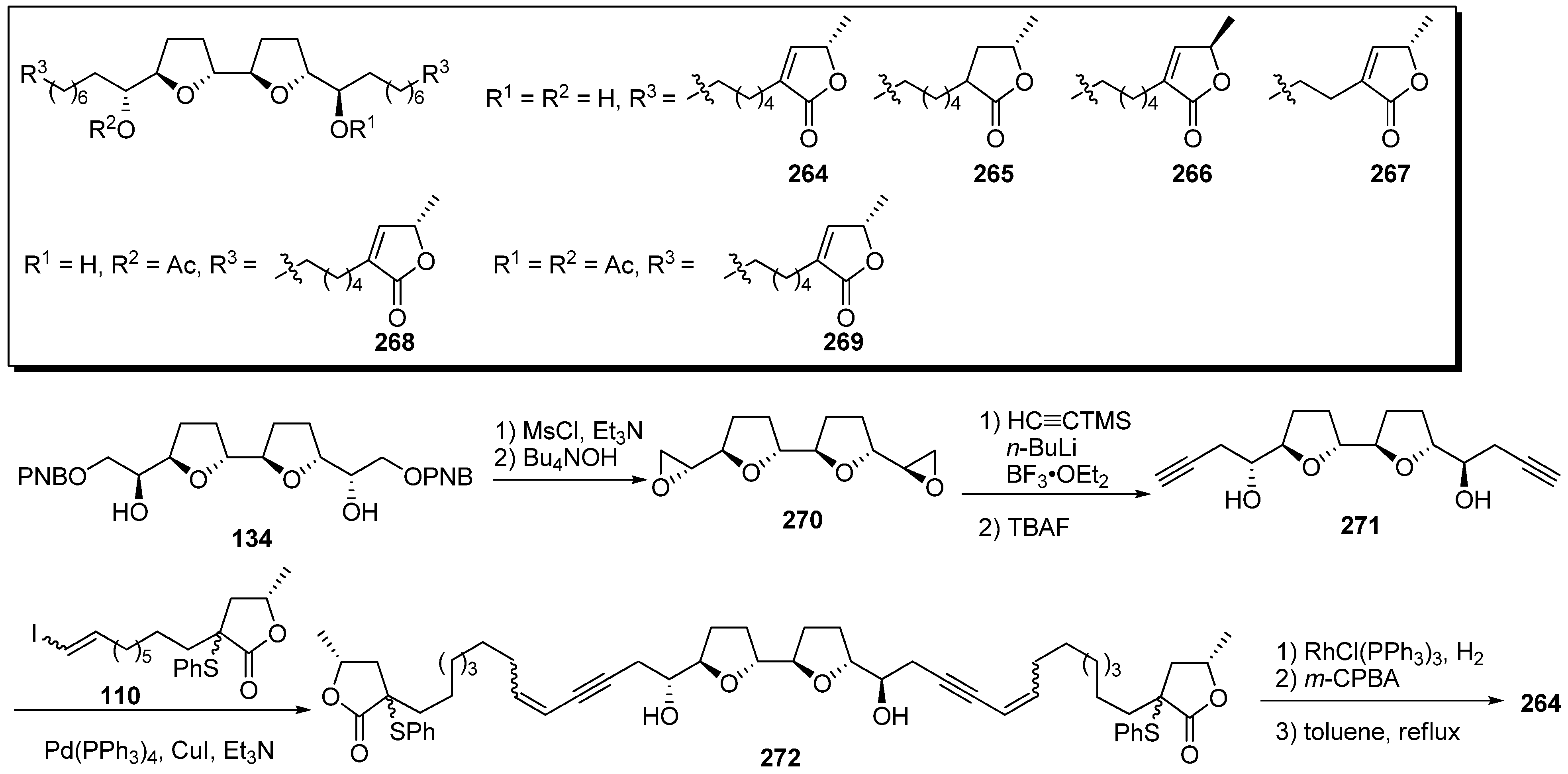
Miyoshi
et al. designed analogues possessing two γ-lactone moieties connected to the bis-THF ring by flexible alkyl chains with the expectation that these analogues would elicit inhibitory activity that was twice as potent as that of ordinary acetogenins [
38], because the γ-lactone moiety was suspected to interact directly with the binding site of complex I. A representative synthetic pathway is given by the preparation of
264 (
Scheme 20). Mesylation of the secondary alcohols of the bis-THF
134, followed by deprotection of the PNB groups, gave bis-epoxide
270. After epoxide opening of
270 with trimethylsilylacetylide, treatment with TBAF afforded diyne
271. The connection of
271 with the γ-lactone fragment
110 was carried out via the Sonogashira reaction. Reduction of
272, followed by the formation of the α,β-unsaturated-γ-lactone moiety, gave the target analogue
264. The inhibitory activities of the synthetic analogues against complex I were measured (
Table 20). The inhibitory activities of analogues
264–
267 were identical to the activity of bullatacin. These results indicate that the analogues
264–
269 does not work as two mole of inhibitors, although they have two mole of γ-lactone moieties in their one molecule, suggesting in turn that one γ-lactone and one THF moieties act cooperatively on the enzyme.
Scheme 20.
Synthesis of analogues possessing two γ-lactone moieties connected to the bis-THF ring by flexible alkyl chains.
Scheme 20.
Synthesis of analogues possessing two γ-lactone moieties connected to the bis-THF ring by flexible alkyl chains.
Table 20.
Inhibition of complex I.
Table 20.
Inhibition of complex I.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| 264 | 1.2 | 268 | 2.0 |
| 265 | 1.6 | 269 | 18 |
| 266 | 1.2 | bullatacin | 1.2 |
| 267 | 1.9 | | |
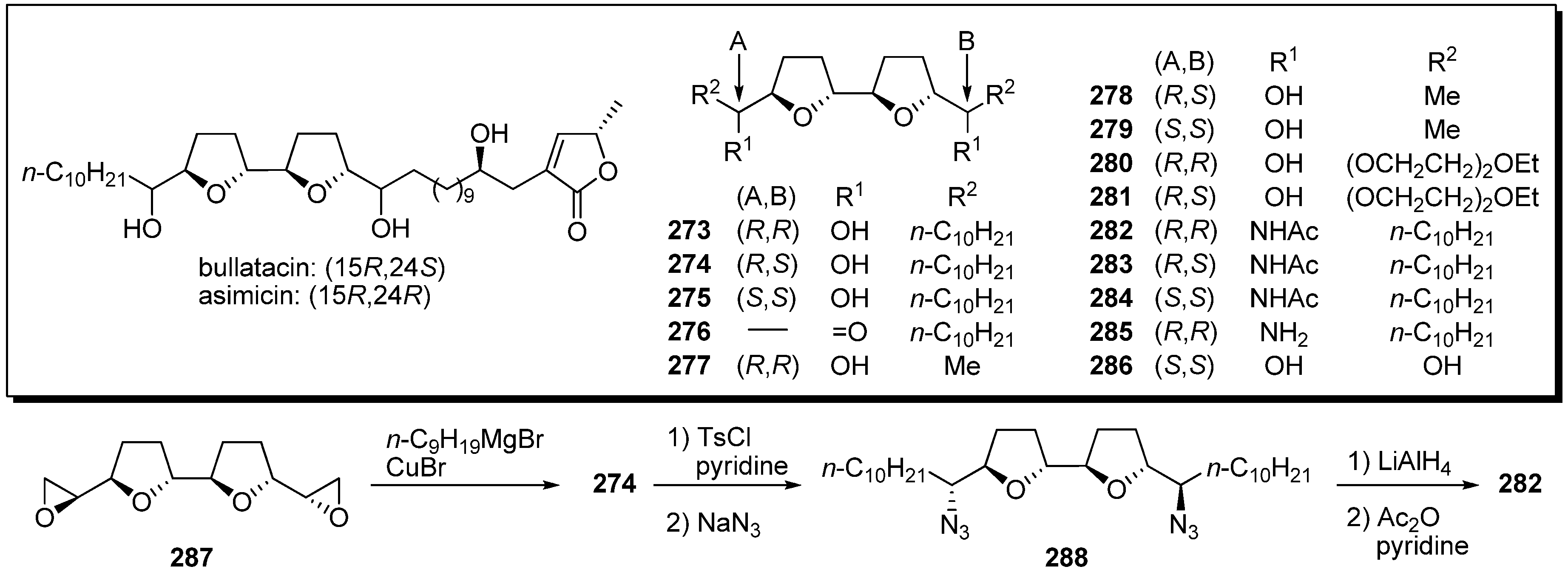
Sasaki and Maeda
et al. tested the assertion that acetogenins possessed affinity toward metal cations. Such properties may be related to their biological activity, because acetogenins are structurally analogous to known ionophores, such as oligo-tetrahydrofurans [
67]. They designed analogues possessing only a bis-THF moiety without the γ-lactone moiety (
Scheme 21) [
25,
26,
68]. A representative synthetic pathway is given by the preparation of
282. Epoxide opening of
287 with a Grignard reagent gave the diol
274. Tosylation of two secondary alcohols followed by azidation afforded the azide
288. After reduction of the azide groups, acetylation of the resulting secondary amines yielded the target analogue
282. Complexation properties of natural acetogenins and analogues were investigated by
1H NMR titration (
Table 21). Among the bis-THF analogues with flanking hydroxyl groups, it was revealed that some analogues (
274 and
277) had selective affinity towards Ca
2+. Very high binding affinity was exhibited by diacetamide
284 to both Mg
2+ and Ca
2+ in the formation of 2:1 ligand-to-metal complexes.
Scheme 21.
Synthesis of analogues without the γ-lactone moiety by Sasaki’s group.
Scheme 21.
Synthesis of analogues without the γ-lactone moiety by Sasaki’s group.
Table 21.
Binding properties of the bis-THF ligands.
Table 21.
Binding properties of the bis-THF ligands.
| Compounds | Metal | Compound/metal | Ks (103 M−1) | Compounds | Metal | Compound/metal | Ks (103 M−1) |
|---|
| bullatacin | Ca2+ | 2:1 | 3.10 | 280 | Ca2+ | 1:1 | 0.08 |
| asimicin | Ca2+ | 4:1 | 5.50 | 280 | Mg2+ | 1:1 | 0.62 |
| 273 | Ca2+ | 4:1 | 0.15 | 281 | Ca2+ | 1:1 | 0.06 |
| 273 | Mg2+ | 4:1 | 0.09 | 281 | Mg2+ | 1:1 | 0.11 |
| 273 | K+ | 4:1 | 0.13 | 282 | Ca2+ | 4:1 | 0.66 |
| 273 | Na+ | 4:1 | 0.04 | 282 | Mg2+ | 2:1 | 4.80 |
| 274 | Ca2+ | 4:1 | 1.50 | 282 | K+ | 4:1 | 0.25 |
| 274 | Mg2+ | 4:1 | 0.21 | 282 | Na+ | 4:1 | 0.28 |
| 275 | Ca2+ | 4:1 | 0.10 | 283 | Ca2+ | 4:1 | 9.60 |
| 277 | Ca2+ | 2:1 | 9.00 | 283 | Mg2+ | 4:1 | 3.00 |
| 277 | Mg2+ | 2:1 | 0.04 | 283 | K+ | 2:1 | 1.00 |
| 277 | K+ | – | – | 283 | Na+ | 4:1 | 1.20 |
| 277 | Na+ | – | – | 284 | Ca2+ | 2:1 | > 100 |
| 278 | Ca2+ | 2:1 | 0.83 | 284 | Mg2+ | 2:1 | > 100 |
| 278 | Mg2+ | 2:1 | 0.06 | 284 | K+ | 4:1 | 1.60 |
| 278 | K+ | – | – | 284 | Na+ | 4:1 | 3.40 |
| 278 | Na+ | – | – | | | | |
| 279 | Ca2+ | 2:1 | 0.05 | | | | |
| 279 | Mg2+ | 2:1 | 0.07 | | | | |
| 279 | K+ | – | – | | | | |
| 279 | Na+ | – | – | | | | |
The GI
50 values of their analogues against cancer cell lines are listed in
Table 22 [
69]. Although the analogues possessing short alkyl chains
277–
279, ether chain
280–
281, or no chain,
286, did not show activity, the analogues
273–
275 with long alkyl chains retained cytotoxicity against P388 cells. The analogues (
282–
283 and
285) with amino groups instead of hydroxyl groups also showed inhibitory activity against cancer cell lines.
Table 22.
GI50 (μM) values against cancer cell lines.
Table 22.
GI50 (μM) values against cancer cell lines.
| Compounds | P388a | PC-6b | NUGC-3c | Compounds | P388a | PC-6b | NUGC-3c |
|---|
| bullatacin | 1.04 × 10−4 | > 0.250 | > 0.250 | 278 | 11.6 | > 50.0 | − |
| asimicin | 3.51 × 10−4 | > 0.250 | > 0.250 | 279 | > 25.0 | > 25.0 | > 25.0 |
| 273 | 0.271 | 6.34 | – | 280 | > 50.0 | > 50.0 | > 50.0 |
| enantio-273 | 1.42 | 16.4 | – | 281 | 35.9 | > 50.0 | > 50.0 |
| 274 | 0.111 | 34.6 | – | 282 | 1.82 | > 2.50 | > 2.50 |
| enantio-274 | 3.10 | > 25.0 | – | 283 | 0.460 | > 2.50 | > 2.50 |
| 275 | 0.140 | 21.9 | – | 284 | > 2.50 | > 2.50 | > 2.50 |
| 276 | > 5.00 | – | – | 285 | 0.610 | 0.484 | 0.722 |
| 277 | > 2.50 | > 25.0 | > 2.50 | 286 | 17.2 | 28.0 | – |
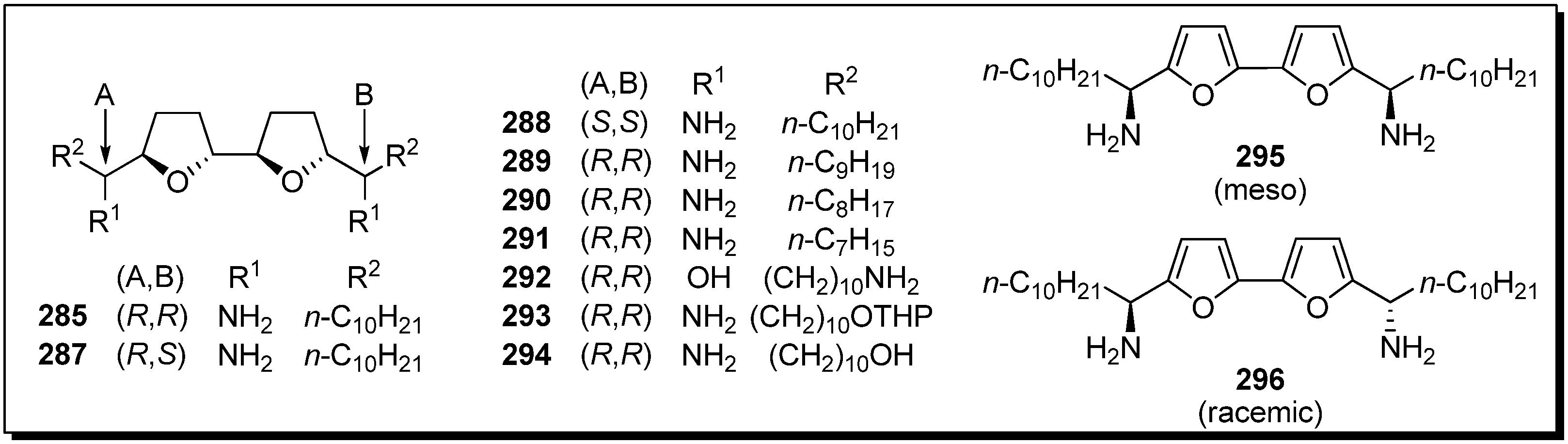
Sasaki
et al. tested the possibility that their bis-THF analogues were active DNA binding agents, because a helix-like conformation was suggested in the studies of naturally occurring bis-THF acetogenins [
70,
71]. The DNA binding affinities of bis-THF analogues were evaluated by Sasaki
et al. (
Figure 3) [
72]. It was revealed that the stereochemistries around the bis-THF moiety and the length of alkyl chains affected the binding affinity. The bis-furan
295 also displayed high affinity against CA12.
Figure 3.
Diamino-bis-THF analogues by Sasaki’s group.
Figure 3.
Diamino-bis-THF analogues by Sasaki’s group.
Table 23.
Comparison of DNA binding affinities.a
Table 23.
Comparison of DNA binding affinities.a
| Compounds | C50 (μM) | Compounds | C50 (μM) |
| CT12b | CA12 c | CT12b | CA12c |
| 285 | 7 | 5.5 | 292 | 300 | 330 |
| 287 | 19 | 13 | 293 | 32 | 27 |
| 288 | 30 | 30 | 294 | 490 | 540 |
| 289 | 34 | 23 | 295 | 17 | 4 |
| 290 | 135 | 125 | 296 | > 100 | > 100 |
| 291 | 760 | 880 | distamycin | 19 | > 15 |
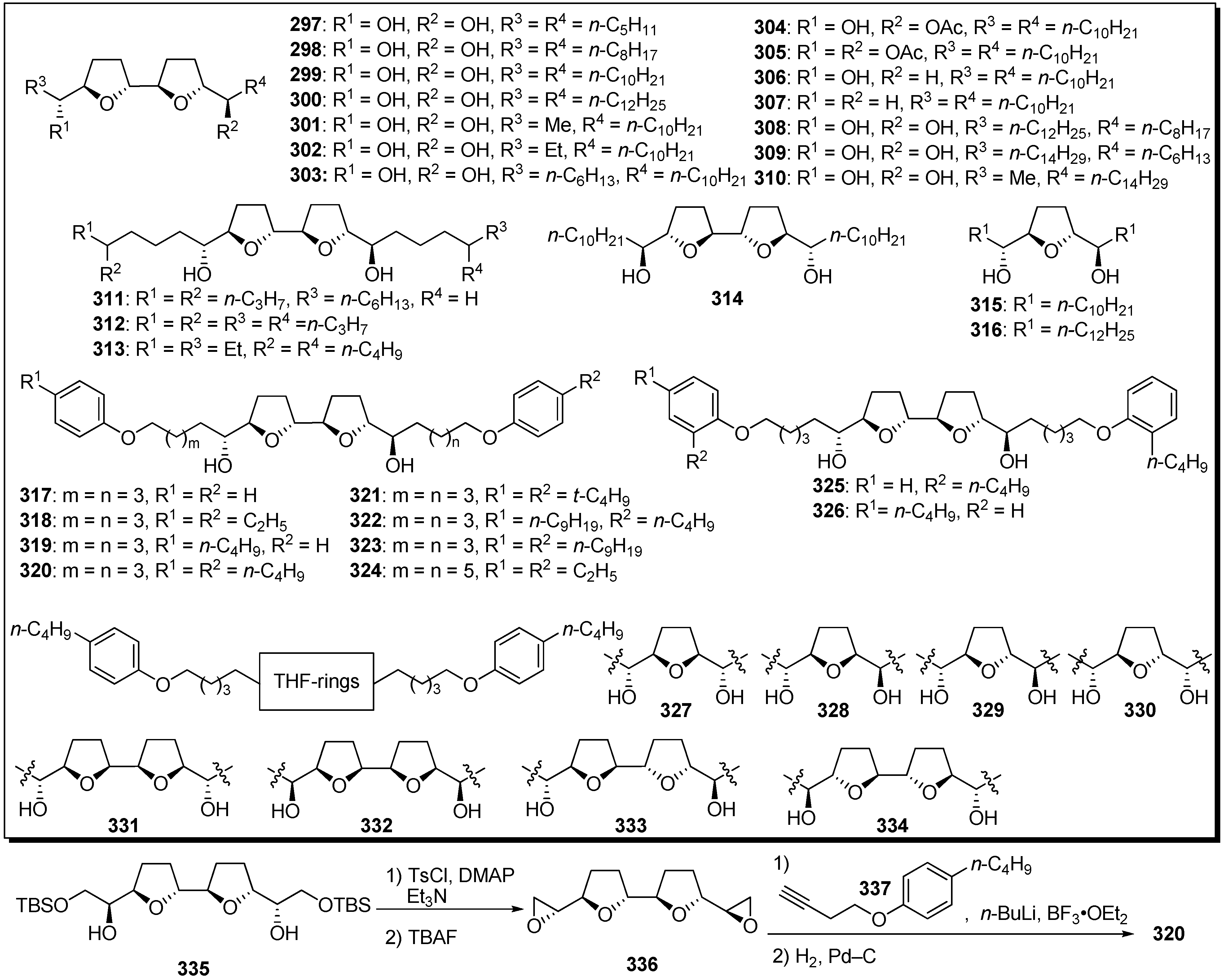
Surprisingly, Miyoshi
et al. discovered that the analogues that possessed two alkyl tails without a γ-lactone in bis-THF acetogenins showed potent inhibitory activity against mitochondrial complex I (
Scheme 22) [
73,
74,
75,
76,
77,
78,
79]. A representative synthetic pathway is given by the preparation of
320. Tosylation of secondary alcohols of bis-THF cores
335, followed by treatment with TBAF, gave the bis-epoxide
336. Epoxide opening of
336 with the acetylide generated from
337, followed by hydrogenation, afforded the target analogue
320. Inhibitory activities of synthetic analogues against complex I were examined (
Table 24). The inhibitory potencies were affected by the length of the alkyl tails, and the analogue
299, possessing unbranched decyl groups, showed the most potent activity among analogues
297–
314. Analogues
317–
321, possessing a phenol moiety at the end of the alkyl chains, exhibited more potent inhibitory activity. It was revealed that the stereochemistry around the THF ring moiety significantly influenced the inhibitory effect of the analogues. A mode-of-action study suggested that the binding site of the analogues is not identical to the binding site of ubiquinone and is downstream of the binding site of ordinary inhibitors.
Scheme 22.
Synthesis of analogues possessing two alkyl chains without γ-lactone by Miyoshi’s group.
Scheme 22.
Synthesis of analogues possessing two alkyl chains without γ-lactone by Miyoshi’s group.
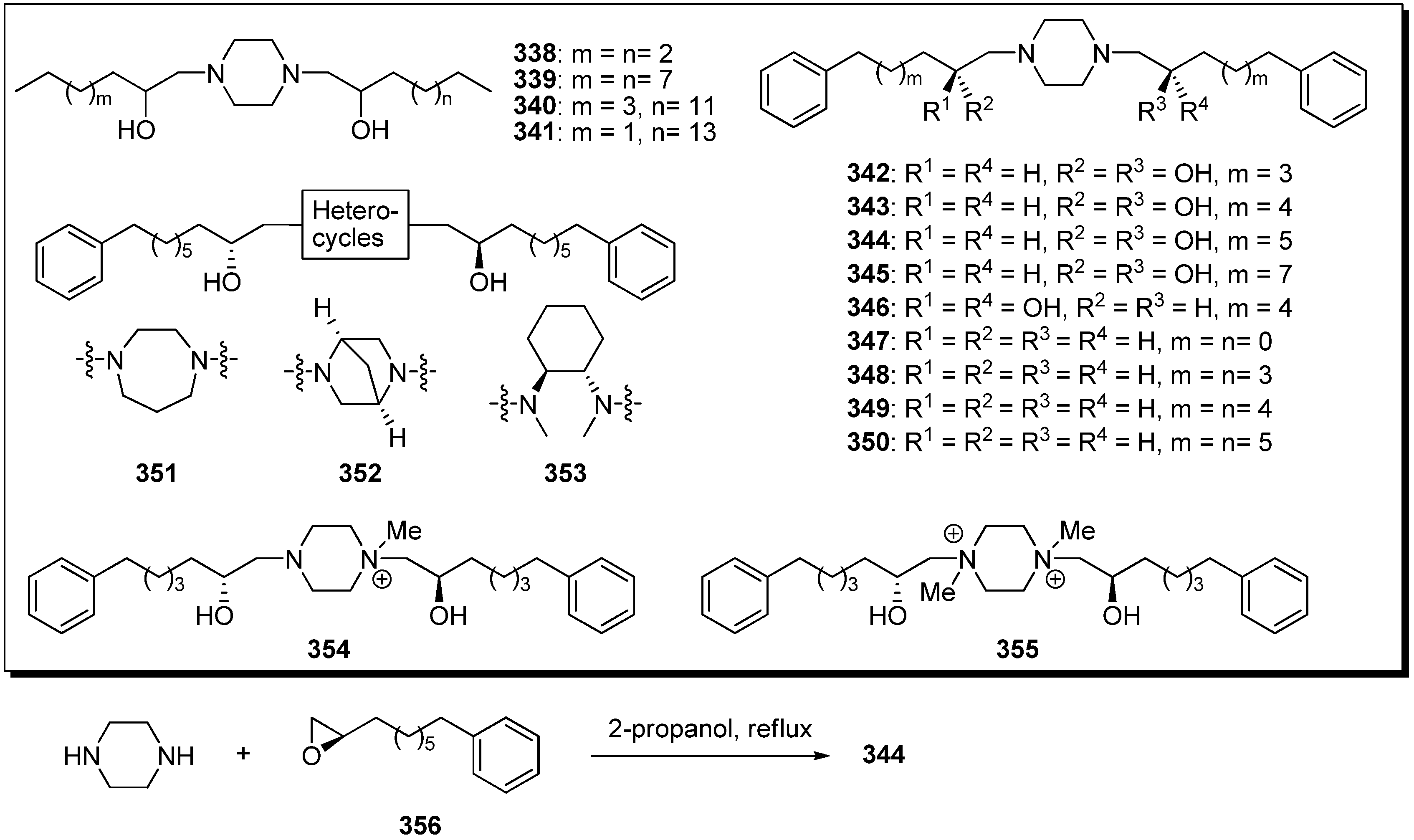
Based on the structural similarities between the hydroxylated bis-THF moiety and the piperazine ring, Miyoshi
et al. designed a series of piperazine derivatives by replacing the bis-THF moiety of their analogues
297–
334 with piperazine rings (
Scheme 23) [
80]. Piperazine analogues (e.g.,
344) were easily prepared by the condensation of piperazine and epoxide
356. The inhibitory activities of synthetic analogues against complex I were examined (
Table 25). Some piperazine analogues (
339,
343–
346) showed potent inhibitory activity equal to that of the parent compounds (e.g.,
320). Although inhibitory potencies were affected by the length of alky chains as well as parent compound, the presence of two hydroxyl groups was not crucial for activity. Modifying the conformational properties of the piperazine rings did not affect activity. The photoaffinity labeling study of new piperazine derivatives revealed that this analogue bound to the 49 kDa subunit and an unidentified subunit (not ND1) with a frequency of ~1:3, but prevented the specific binding of [
125I](trifluoromethyl)phenyldiazirinyl acetogenin to the ND1 subunit.
Table 24.
Summary of the inhibitory potencies against complex I.
Table 24.
Summary of the inhibitory potencies against complex I.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) | Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| 297 | 4500 | 307 | 620 | 317 | 1.4 | 327 | 39 |
| 298 | 45 | 308 | 7.5 | 318 | 1.1 | 328 | 41 |
| 299 | 1.6 | 309 | 34 | 319 | 0.91 | 329 | 47 |
| 300 | 9.0 | 310 | 410 | 320 | 0.83 | 330 | 38 |
| 301 | 280 | 311 | 27 | 321 | 1.0 | 331 | 16 |
| 302 | 45 | 312 | 1500 | 322 | 150 | 332 | 9.0 |
| 303 | 3.2 | 313 | 870 | 323 | > 500 | 333 | 5.2 |
| 304 | 5.5 | 314 | 7.5 | 324 | 3.0 | 334 | 3.8 |
| 305 | 330 | 315 | 308 | 325 | > 1000 | bullatacin | 0.85 |
| 306 | 14 | 316 | > 25000 | 326 | 16 | | |
Scheme 23.
Synthesis of analogues possessing piperazines instead of the bis-THF moiety by Miyoshi’s group.
Scheme 23.
Synthesis of analogues possessing piperazines instead of the bis-THF moiety by Miyoshi’s group.
Table 25.
Summary of the inhibitory potencies against complex I.
Table 25.
Summary of the inhibitory potencies against complex I.
| Compounds | IC50 (nM) | Compounds | IC50 (nM) |
|---|
| 338 | > 22000 | 347 | 1300 |
| 339 | 2.6 | 348 | 12 |
| 340 | 26 | 349 | 2.2 |
| 341 | 110 | 350 | 3.6 |
| 342 | 12 | 351 | 1.7 |
| 343 | 1.7 | 352 | 2.8 |
| 344 | 1.2 | 353 | 670 |
| 345 | 5.9 | 354 | 1100 |
| 346 | 2.3 | 355 | 4700 |
5. Modification of the Oxygenated Moiety on Alkyl Chains
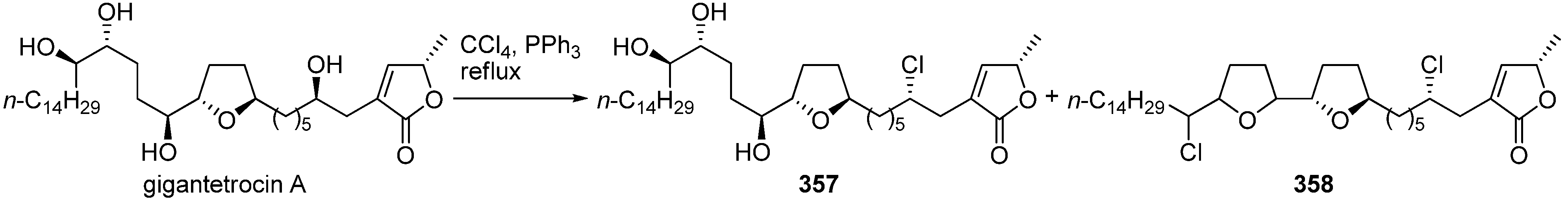
McLaughlin
et al. reported a semisynthesis of chlorinated analogues,
357–
358, of gigantetrocin A (
Scheme 24) [
81]. Gigantetrocin A was refluxed with PPh
3 in CCl
4 to give a mixture of 4-chloro-4-deoxygigantetrocin A
357 and dichloro bis-THF derivative
358. Both chlorinated analogues
357–
358 showed decreased bioactivity against tumor cell lines compared with gigantetrocin A (
Table 26). However, the 4-chloro derivative
357 was selectively cytotoxic to HT-29, and
358 was selectively cytotoxic to PC-3.
Scheme 24.
Semisynthesis of chlorinated acetogenins by McLaughlin’s group.
Scheme 24.
Semisynthesis of chlorinated acetogenins by McLaughlin’s group.
Table 26.
Bioactivity data ofchlorinated analogues 357–358 (LC50 and ED50: μg/mL).
Table 26.
Bioactivity data ofchlorinated analogues 357–358 (LC50 and ED50: μg/mL).
| Compounds | BSTa | A-549b | MCF-7c | HT-29d | A-498e | PC-3f | PaCa-2g |
|---|
| gigantetrocin A | 2.6 | 2.5 × 10−1 | 6.3 × 10−1 | 4.1 × 10−5 | nt | nt | nt |
| 357 | 54.6 | > 10 | 5.74 | 3.8 × 10−1 | > 10 | > 10 | > 10 |
| 358 | 31.9 | 2.39 | > 10 | > 10 | 2.47 | 7.55 × 10−1 | 1.16 |
| adriamycin | nt | 2.43 × 10−2 | 2.09 × 10−1 | 3.46 × 10−2 | 6.49 × 10−3 | 2.62 × 10−2 | 7.90 × 10−3 |
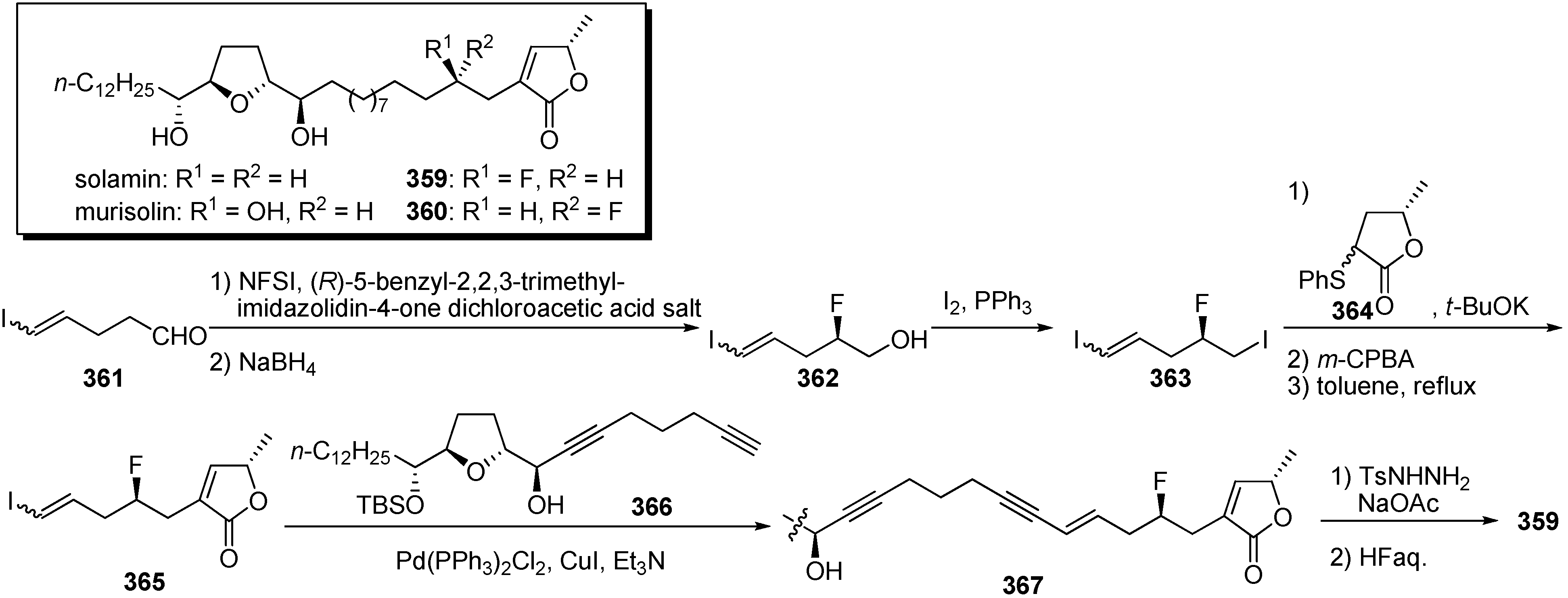
The presence of a C4-hydroxyl group in acetogenins is known to influence cytotoxic activity against tumor cell lines. Kojima and Tanaka
et al. were interested in whether C4-fluorinated solamin showed similar activity to that of solamin or murisolin (C4-(
R)-hydroxyl solamin), because fluorine atoms are known to mimic hydrogen atoms. Substitution of the hydroxyl group with an isoelectronic function such as fluorine in biologically active compounds can produce more potent analogues (
Scheme 25) [
82]. The synthesis of
359 started from enantioselective α-fluorination [
83] of aldehyde
361, followed by reduction of the aldehyde, to give the alcohol
362. After iodination of the primary alcohol of
362, the coupling reaction with the γ-lactone
364 and the sequential thermal elimination of the sulfide moiety afforded α,β-unsaturated γ-lactone
365. Assembly of two fragments,
365 and
366, prepared by a systematic construction strategy [
84,
85,
86,
87,
88,
89,
90,
91,
92], with the Sonogashira reaction, followed by selective reduction of endiyne and deprotection of the TBS ether, gave the target fluorinated analogue
359. Synthetic analogues
359–
360 were tested for
in vitro antiproliferative activity against a panel of 39 human cancer cell lines (
Table 27). Selected results are summarized in
Table 27. Two fluorinated analogues,
359 and
360, displayed inhibitory potency levels that fell between the potencies of solamin and murisolin. Interestingly, the analogue
359 showed approximately 20 times higher cytotoxicity to MKN7 than did
360, implying that the stereochemistry of the fluorine atom at the C4 position was recognized by cancer cell lines.
Scheme 25.
Synthesis of C4-fluorinated solamins by Tanaka’s group.
Scheme 25.
Synthesis of C4-fluorinated solamins by Tanaka’s group.
Table 27.
Selected GI50 (M) values of fluorinated analogues against 39 human cancer cell lines.
Table 27.
Selected GI50 (M) values of fluorinated analogues against 39 human cancer cell lines.
| Compounds | MCF-7a | SF-295b | HCT-116c | NCI-H23d | DMS114d | MKN7e | PC-3f |
| solamin | 7.1 × 10−5 | 4.0 × 10−5 | > 10−4 | 7.3 × 10−5 | 4.3 × 10−6 | 1.3 × 10−5 | > 10−4 |
| murisolin | 3.9 × 10−6 | 7.3 × 10−6 | 3.7 × 10−6 | 2.2 × 10−7 | < 10−8 | 7.0 × 10−7 | 1.7 × 10−5 |
| 359 | 5.6 × 10−5 | 3.7 × 10−5 | 2.0 × 10−6 | 2.3 × 10−5 | 2.6 × 10−7 | 9.6 × 10−7 | > 10−4 |
| 360 | 3.7 × 10−5 | 3.1 × 10−5 | 2.7 × 10−5 | 5.1 × 10−6 | 4.6 × 10−7 | 1.9 × 10−6 | 8.4 × 10−5 |
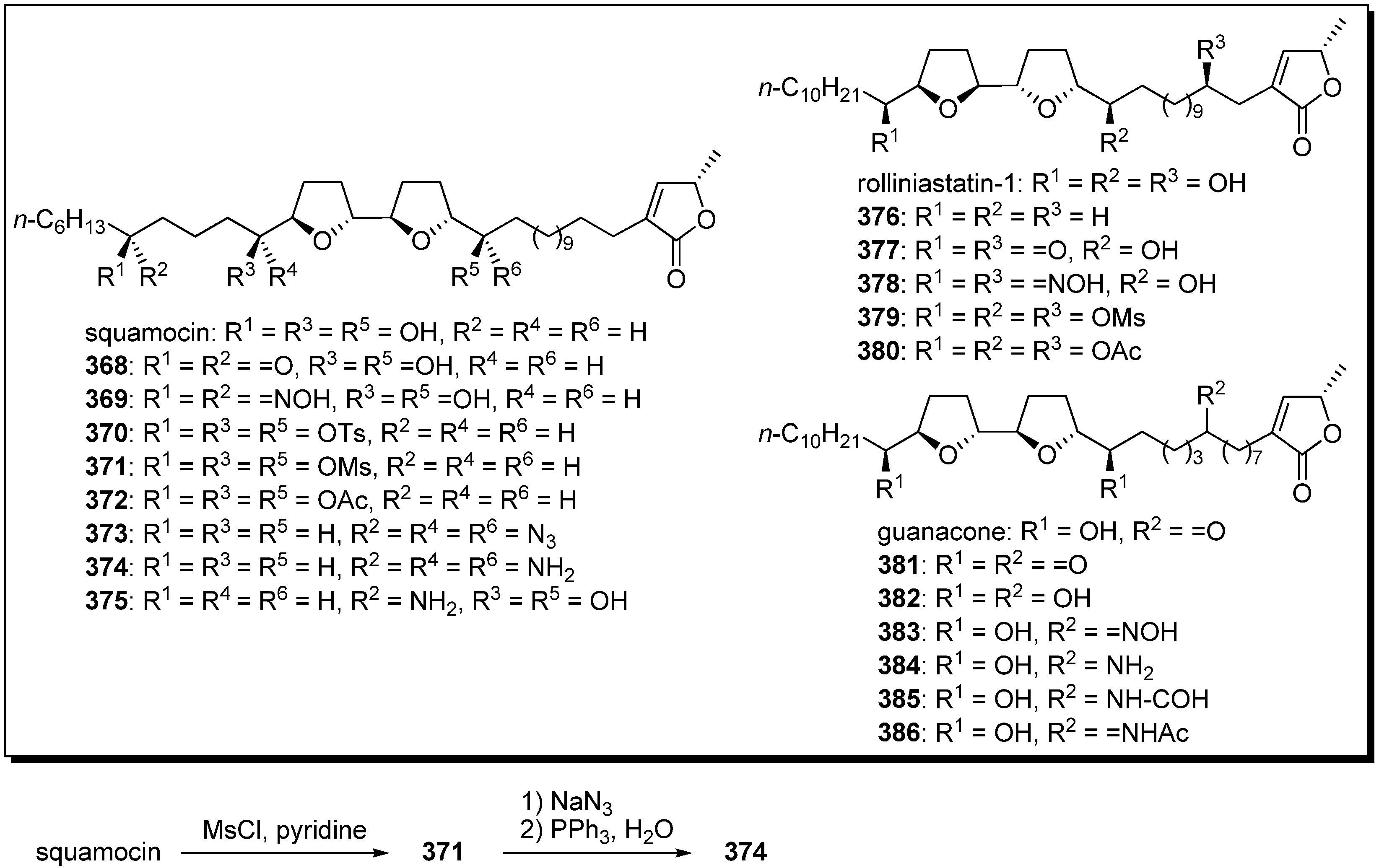
Semisynthesis of analogues possessing substituted functionalities in place of the hydroxyl groups were independently reported by Cortes’s group [
53,
93,
94,
95] and Figadère’s group [
96].
A representative synthetic pathway is given by the preparation of
374. Treatment of squamocin with mesyl chloride in pyridine gave the mesylate
371. Azidation of
371, followed by Staudinger reduction, afforded the target analogue
374. The inhibitory potency of analogues against complex I were examined by Cortes [
53,
93,
94,
95]. The oxo and hydroxyimino derivatives showed potent activity, but the activities of acetyl, mesyl, and azide analogues were diminished. The cytotoxicities of analogues against cancer cell lines were evaluated by Figadère
et al. [
96]. All of the tested squamocin analogues exhibited much lower cytotoxicity than did natural products. On the other hand, Cortes’s group reported that guanacone analogus
381–
386 showed more potent cytotoxicity against human cancer cell lines than parent compound [
94,
95].
Scheme 26.
Semisynthesis of acetogenin analogues modified by an oxygenated moiety on the alkyl chains by Cortes’ group and Figadère’s group.
Scheme 26.
Semisynthesis of acetogenin analogues modified by an oxygenated moiety on the alkyl chains by Cortes’ group and Figadère’s group.
Table 28.
Inhibitory potency against complex I and cytotoxicity against cancer cell lines.
Table 28.
Inhibitory potency against complex I and cytotoxicity against cancer cell lines.
| Compounds | IC50 (nM) NADH oxidase | EC50 (μM) | Compounds | IC50 (nM) NADH oxidase | EC50 (μM) |
| KB | VERO | KB | VERO |
| squamocin | 0.59 | 1.6 × 10−5 | 4.8 × 10−2 | rolliniastatin-1 | 0.60 | < 1.6 × 10−7 | 1.1 × 10−2 |
| 368 | 0.65 | nt | nt | 376 | nt | 3.5 × 10−1 | 1.4 × 10−1 |
| 369 | 0.74 | nt | nt | 377 | 0.34 | nt | nt |
| 370 | nt | < 1.3 × 10−1 | < 1.3 × 10−1 | 378 | 0.34 | nt | nt |
| 371 | 14.1 | 1.5 × 10−2 | < 10−1 | 379 | 1.68 | nt | nt |
| 372 | 5.0 | nt | nt | 380 | 1.46 | nt | nt |
| 373 | 18 | < 1.4 × 10−1 | < 1.4 × 10−1 | guanacone | 1.52 | nt | nt |
| 374 | nt | 2.4× 10−2 | < 1.6 × 10−1 | 381 | 1.65 | nt | nt |
| 375 | nt | 9.7 × 10−2 | < 1.6 × 10−1 | 382 | 0.95 | nt | nt |
| | | | | 383 | 0.34 | nt | nt |
| | | | | 384 | 12.8 | nt | nt |
| | | | | 385 | 5.5 | nt | nt |
| | | | | 386 | nt | nt | nt |
Table 28 shows that the transformation of the secondary alcohols into acetate or mesylate led to a disappearance of cytotoxicity, but such derivatives retained strong activity against mitochondrial complex I.
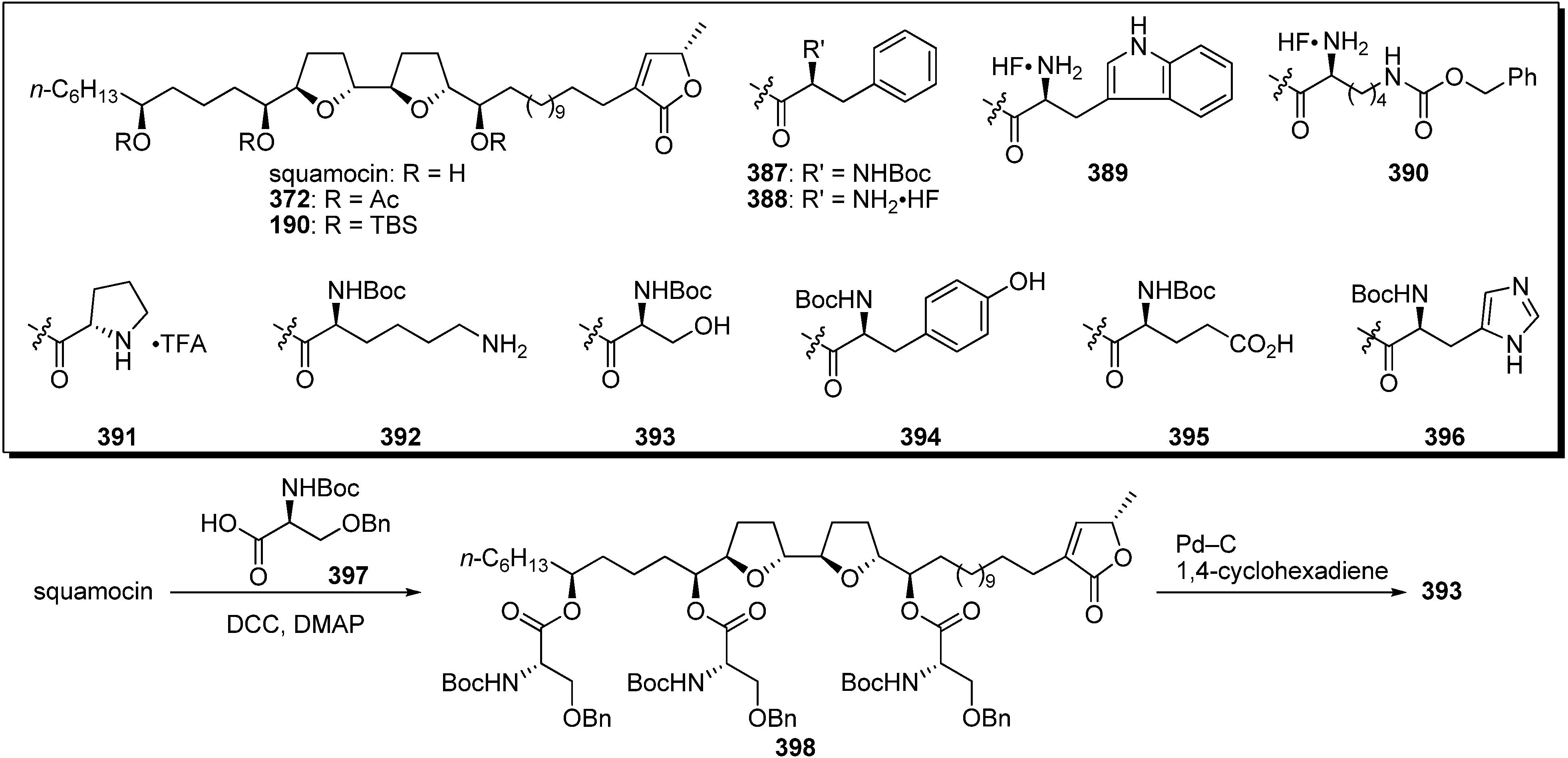
Scheme 27.
Semisynthesis of aminoacyl triester of squamocin by Lewin’s group.
Scheme 27.
Semisynthesis of aminoacyl triester of squamocin by Lewin’s group.
To explain this ambiguity, Lewin
et al. carried out semisynthesis and evaluation of biological activities of aminoacyl squamocin (
Scheme 27) [
97]. A representative synthetic pathway is given by the preparation of
393. The esterification of squamocin with the
N-boc-protected serine
397 gave the triester
398. Deprotection of tri-benzyl ether using Pd–C/1,4-cyclohexadiene system afforded the target analogue
393. The inhibitory activity against complex I and cytotoxicity against KB 3-1 cell lines were examined (
Table 29). Despite enhanced polarity compared with the natural acetogenins, all derivatives showed strongly reduced activities, although their analogues were more strongly active than was the lipophilic trisilyl ether
190.
Table 29.
Inhibitory potency against complex I and cytotoxicity against cancer cell lines.
Table 29.
Inhibitory potency against complex I and cytotoxicity against cancer cell lines.
| Compounds | IC50 (nM) NADH oxidase | IC50 (nM) | Compounds | IC50 (nM) NADH oxidase | IC50 (nM) |
|---|
| KB 3-1 | KB 3-1 |
|---|
| squamocin | 0.8 | 2.6 × 10−14 | 391 | 247.7 | 2.0 × 10−8 |
| 372 | 5.0 | nt | 392 | nt | 1.1 × 10−6 |
| 190 | > 5000 | > 10−5 | 393 | nt | 5.5 × 10−7 |
| 387 | 625 | nt | 394 | nt | > 10−5 |
| 388 | 440.0 | 6.1 × 10−8 | 395 | nt | > 10−5 |
| 389 | nt | 2.4 × 10−7 | 396 | nt | 1.7 × 10−6 |
| 390 | nt | 6.3 × 10−8 | | | |
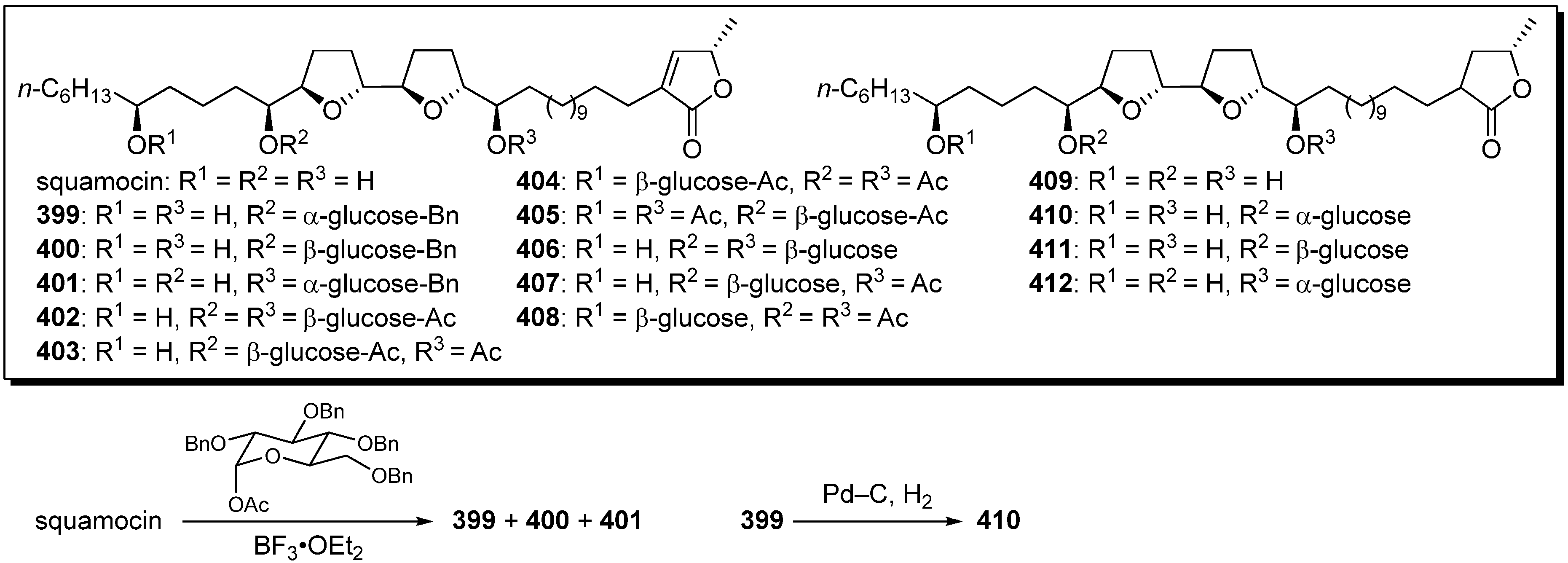
To investigate the influence of aqueous solubility of acetogenins on their cytotoxicity, a series of glycosyl analogues of squamocin was synthesized by Figadère’s group (
Scheme 28) [
98]. A representative synthetic pathway is given by the preparation of
410. Lewis acid catalyzed glycosylation of squamocin with 1-acetyl-2,3,4,6-tetrabenzyl-α-
D-glucopyranose gave a mixture of
399-
401. After purification by HPLC, hydrogenation of the compound
399 gave target analogue
410. Cytotoxicity against KB, VERO, and L1210 cell lines were examined (
Table 30). Glycosylated analogues showed low activity against the normal cell (VERO) while still active against cancer cell lines (KB and L1210). The water solubility diminished the cytotoxicity probably because of low lipophilicity to cross the cell membrane. Interestingly, two analogues
401 and
411 have shown significant inhibition of the proliferation of L1210 in the G1 phase, whereas squamocin was not specific.
Scheme 28.
Semisynthesis of glycosyl analogues of squamocin by Figadère’s group.
Scheme 28.
Semisynthesis of glycosyl analogues of squamocin by Figadère’s group.
Table 30.
Cytotoxic activity of glycosyl analogues of squamocin by Figadère’s group.
Table 30.
Cytotoxic activity of glycosyl analogues of squamocin by Figadère’s group.
| Compounds | EC50 (μM) | Compounds | EC50 (μM) |
| KBa | VEROb | L1210c | KBa | VEROb | L1210c |
| squamocin | 1.6 × 10−5 | 1.6 × 10−2 | < 4.0 × 10−4 | 408 | 1.1 × 10−2 | 3.4 × 10−1 | 1.7 × 10−2 |
| 399 | 4.3 × 10−1 | 8.7 × 10−1 | nt | 409 | 2.4 × 10−4 | 1.6 × 10−2 | < 2.5 × 10−4 |
| 400 | nt | nt | 5.0 × 10−2 | 410 | 1.9 × 10−3 | 6.0 × 10−1 | 1.0 × 10−2 |
| 404 | 9.6 × 10−4 | 2.0 × 10−2 | 3.0 × 10−1 | 411 | nt | nt | 8.0 × 10−2 |
| 406 | 1.05 | 1.05 | 10 | vinblastine | 1.2 × 10−3 | < 3.7 | nt |
| 407 | 3.6 × 10−1 | 1.21 | 50 | | | | |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




























