Synthesis of Novel Asymmetric Zinc (II) Phthalocyanines Bearing Octadecyloxyl and Glucosyl Groups

{kind=link}
{kind=link}
Abstract
:1. Introduction
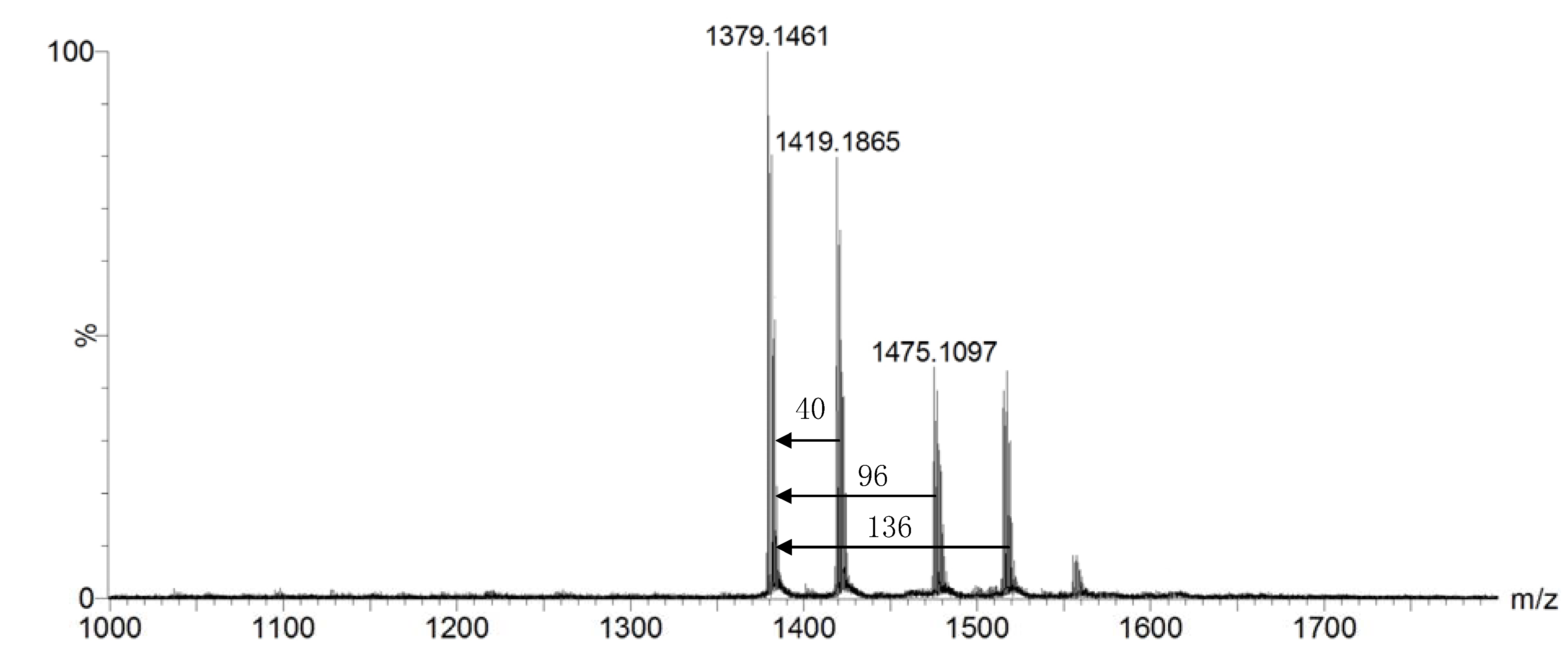
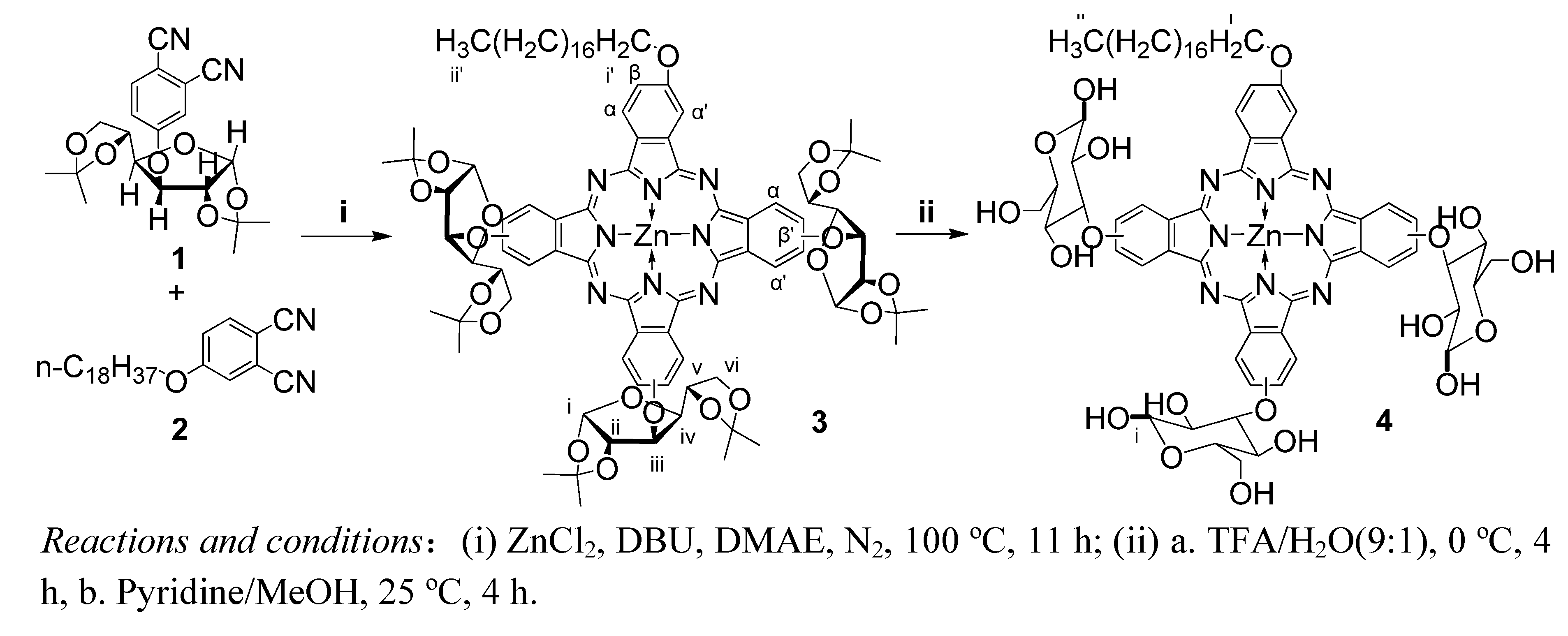
2. Results and Discussion


3. Experimental
3.1. General methods
3.2. Synthesis of [2(3),9(10),16(17)-tris(1,2:5,6-di-O-isopropylidene-a-D-glucofuranosyl)-23(24)-octadecyloxyl phthalocyaninato] zinc (II) (3)
3.3. Synthesis of [2(3), 9(10), 16(17)-tris(glucosyl)-23(24)-octadecyloxyl phthalocyaninato] zinc (II) (4)
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds are available from the authors.
References and Notes
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Ochsner, M. Photophysical and photobiological processes in the photodynamic therapy of tumors. J. Photochem. Photobiol. B 1997, 39, 1–18. [Google Scholar] [CrossRef]
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer I 1998, 90, 889–905. [Google Scholar] [CrossRef]
- His, R.A.; Rosenthal, D.I.; Glastein, E. Photodynamic therapy in the treatment of cancer: Current state of the art. Drugs 1999, 57, 725–734. [Google Scholar] [CrossRef]
- Adams, K.R.; Berembaum, M.C.; Bonnett, R.; Nizhnik, A.N.; Salgado, A. Second generation tumour photosensitisers: The synthesis and biological activity of octaalkyl chlorins and bacteriochlorins with graded amphiphilic character. J. Chem. Soc. Perkin Trans. 1 1992, 1465–1470. [Google Scholar]
- Bonnett, R.; Nizhnik, A.N.; White, R.D.; Berembaum, M.C. Porphyrin sensitizers in tumor phototherapy. Novel sensitizers of the chlorin and bacteriochlorin class with amphiphilic properties. J. Photochem. Photobiol. B 1990, 6, 29–37. [Google Scholar] [CrossRef]
- Bonnett, R. Photodynamic Action. In Advanced Chemistry Texts Chemical Aspects of Photodynamic Therapy; Gordon and Breach Science Publishers: London, UK, 2000; Volume 1, pp. 130–147. [Google Scholar]
- Zheng, G.; Potter, W.R.; Camacho, S.H.; Missert, J.R.; Wang, G.; Bellnier, D.A.; Henderson, B.W.; Rodgers, M.A.J.; Dougherty, T.J.; Pandey, R.K. Synthesis, photophysical properties, tumor uptake, and preliminary in vivo photosensitizing efficacy of a homologous series of 3-(1’-Alkyloxy)ethyl-3-devinylpurpurin-18-N-alkylimides with variable lipophilicity. J. Med. Chem. 2001, 44, 1540–1559. [Google Scholar] [CrossRef]
- Bonnet, R.; Martinez, G. Photobleaching of sensitizers used in photodynamic therapy. Tetrahedron 2001, 57, 9513–9547. [Google Scholar] [CrossRef]
- Macdonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porphyr. Phthalocya 2001, 5, 105–129. [Google Scholar] [CrossRef]
- Ogunsipe, A.; Nyokong, T. Photophysical and photochemical studies of sulphonated non-transition metal phthalocyanines in aqueous and non-aqueous media. J. Photochem. Photobiol. A 2005, 173, 211–220. [Google Scholar] [CrossRef]
- Brown, R.S.; Wahl, R.L. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer 1993, 72, 2979–2985. [Google Scholar] [CrossRef]
- Maillard, P.; Guerquin-Kern, J.L.; Momenteau, M. Glycoconjugated tetrapyrrolic macrocycles. J. Am. Chem. Soc. 1989, 111, 9125–9127. [Google Scholar] [CrossRef]
- Lee, P.P.S.; Lo, P.C.; Chan, E.Y.M.; Fong, W.P.; Ko, W.H.; Ng, D.K.P. Synthesis and in vitro photodynamic activity of novel galactose containing phthalocyanines. Tetrahedron Lett. 2005, 46, 1551–1554. [Google Scholar] [CrossRef]
- Alvarez, M.X.; Calvete, M.J.F.; Hanack, M.; Ziegler, T. The first example of anomeric glycoconjugation to phthalocyanines. Tetrahedron Lett. 2006, 47, 3283–3286. [Google Scholar] [CrossRef]
- Ribeiro, A.O.; Tomé, J.P.C.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Iamamotob, Y.; Torres, T. [1,2,3,4-Tetrakis(α/β-D-galactopyranos-6-yl)phthalocyaninato]zinc(II): A water-soluble phthalocyanine. Tetrahedron Lett. 2006, 47, 9177–9180. [Google Scholar]
- Xavier, Á.M.; Mario, J.F.C.; Michael, H.; Thomas, Z. Expeditious Synthesis of Glycosylated Phthalocyanines. Synthesis 2007, 14, 2186–2192. [Google Scholar]
- Zafar, I.; Michael, H.; Thomas, Z. Synthesis of an octasubstituted galactose zinc(II) phthalocyanine. Tetrahedron Lett. 2009, 50, 873–875. [Google Scholar] [CrossRef]
- Ufuk, K.; Mahmut, A.E.; Fabienne, D.; Vefa, A. Amphiphilic galactosylated phthalocyanines. J. Porphyr. Phthalocya 2008, 12, 1090–1095. [Google Scholar] [CrossRef]
- Drain, C.M.; Christensen, B.; Mauzerall, D.C.P. Photogating of ionic currents across a lipid bilayer. Proc. Natl. Acad. Sci. USA 1998, 86, 6959–6962. [Google Scholar]
- Drain, C.M.; Mauzerall, D.C. Photogating of ionic currents across lipid bilayers. Hydrophobic ion conductance by an ion chain mechanism. Biophys. J. 1992, 63, 1556–1563. [Google Scholar] [CrossRef]
- Tomoda, H.; Asito, S. Phosphonates and thiophosphonates as sulfate surrogates: synthesis of estrone 3-methylthiophosphonate, a potent inhibitor of estrone sulfatase. Chem. Lett. 1983, 3, 313–318. [Google Scholar] [CrossRef]
- Choi, C.F.; Huang, J.D.; Lo, P.C.; Fong, W.P.; Ng., D.K.P. Glycosylated zinc(II) phthalocyanines as efficient photosensitisers for photodynamic therapy. Synthesis, photophysical properties and in vitro photodynamic activity. Org. Biomol. Chem. 2008, 6, 2173–2181. [Google Scholar] [CrossRef]
- Takatani, M.; Matsuo, I.; Ito, Y. Pentafluoropropionyl and trifluoroacetyl groups for temporary hydroxyl group protection in oligomannoside synthesis. Carbohydr. Res. 2003, 338, 1073–1081. [Google Scholar] [CrossRef]
- Chauhan, S.; Kumari, P.; Agarwal, S. Efficient synthesis of transition-metal phthalocyanines in functional ionic liquids. Synthesis 2007, 23, 3713–3721. [Google Scholar]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, P.; Zhang, S.; Han, G. Synthesis of Novel Asymmetric Zinc (II) Phthalocyanines Bearing Octadecyloxyl and Glucosyl Groups. Molecules 2009, 14, 3688-3693. https://doi.org/10.3390/molecules14093688
Zhang P, Zhang S, Han G. Synthesis of Novel Asymmetric Zinc (II) Phthalocyanines Bearing Octadecyloxyl and Glucosyl Groups. Molecules. 2009; 14(9):3688-3693. https://doi.org/10.3390/molecules14093688
Chicago/Turabian StyleZhang, Pei, Shufen Zhang, and Gang Han. 2009. "Synthesis of Novel Asymmetric Zinc (II) Phthalocyanines Bearing Octadecyloxyl and Glucosyl Groups" Molecules 14, no. 9: 3688-3693. https://doi.org/10.3390/molecules14093688
APA StyleZhang, P., Zhang, S., & Han, G. (2009). Synthesis of Novel Asymmetric Zinc (II) Phthalocyanines Bearing Octadecyloxyl and Glucosyl Groups. Molecules, 14(9), 3688-3693. https://doi.org/10.3390/molecules14093688