Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation

Abstract
:
Abbreviations
| Ac | acetyl |
| ACN | acetonitrile |
| Ac2O | acetic anhydride |
| aq. | aqueous |
| 9-BBN | 9-borabicyclo[3.3.1]nonane |
| Bn | benzyl |
| Boc2O | di-tert-butyl dicarbonate |
| cat. | catalyst |
| CuTC | copper(I)-thiophene-2-carboxylate |
| Cy | cyclohexyl |
| CM | cross metathesis |
| DCC | dicyclohexylcarbodiimide |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| de. | diastereomeric excess |
| DIPT | diisopropyl tartrate |
| DMAP | 4-dimethylaminopyridine |
| DME | dimethoxyethane |
| DMF | dimethylformamide |
| DMP | Dess-Martin periodinane: 1,1,1-triacetoxy-1,1-dihydro-1,2-benziodoxol-3(1H)-one |
| DMPM | 3,4-dimethoxybenzyl |
| DMSO | dimethyl sulfoxide |
| dr | diastereomeric ratio |
| ee | enantiomeric excess |
| HMPA | hexamethylphosphoramide |
| Imid. | imidazole |
| Mes | 2,4,6-trimethylphenyl |
| MS | molecular sieves |
| MsCl | mesityl chloride |
| NMO | N-methyl-morpholine-N-oxide |
| nm | not measured |
| Pip. | piperidine |
| PMB | p-methoxybenzyl |
| PSA | p-TsOH.H2O: p-toluenesulfonic acid monohydrate |
| Pyr. | pyridine |
| RedAl | sodium bis(2-methoxyethoxy)aluminum hydride |
| RCM | ring-closing metathesis |
| SAE | Sharpless asymmetric epoxidation |
| TASF | tris(dimethylamino)sulfonium difluorotrimethylsilicate |
| TBAF | tetra-n-butylammonium fluoride |
| TBS | tert-butyldimethylsilyl |
| TBDPS | tert-butyldiphenylsilyl |
| TEMPO | 2,2,6,6-tetramethylpiperidine-1-oxyl |
| TES | triethylsilyl |
| Tf | triflate |
| THF | tetrahydrofuran |
| TIPS | triisopropylsilyl |
| TMS | trimethylsilyl |
| TPAP | tetrapropylammonium perruthenate |
| TsCl | m-toluenesulfonyl chloride |
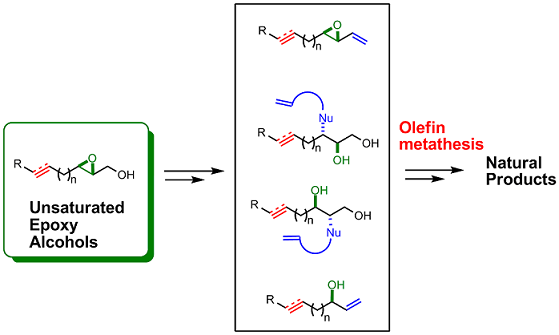
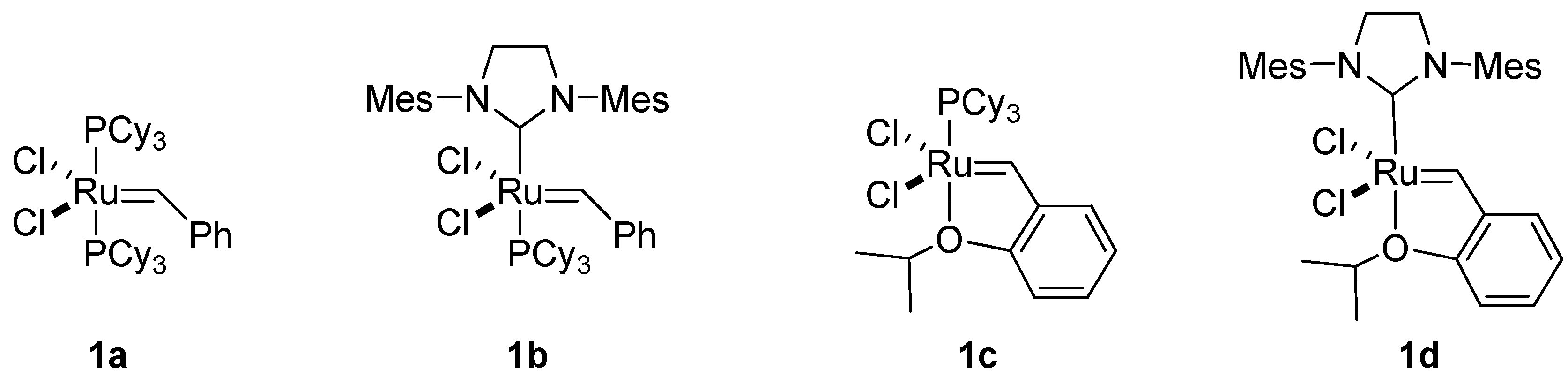
1. Introduction


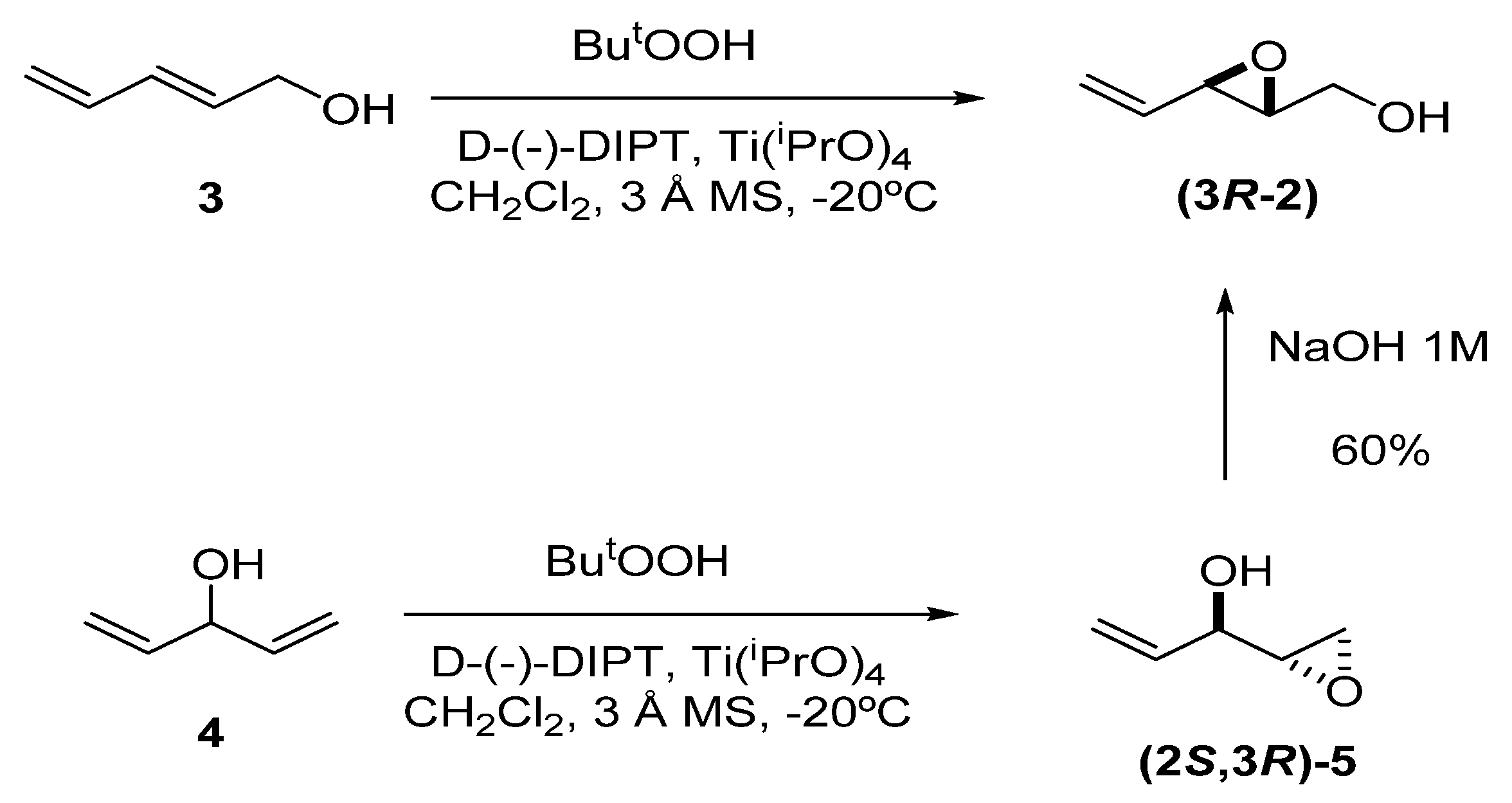
2. Synthesis of Chiral Epoxy Alcohols


| Epoxy alcohol | Structure | Yield (%) | ee (%) | References |
|---|---|---|---|---|
| 2 |  | n.d.* | 96 | [13,57,58,85] |
| 6 |  | 50 | nd | [74] |
| 7 |  | 98 | 61 | [53] |
| 8 |  | 83 | 88 | [52] |
| 9 |  | 85 | 95 | [51] |
| 10 |  | 21** | 96 | [51] |
| 11 |  | 82 | 92 | [82],[47] |
| Z 11 |  | 82 | >99 | [28],[45] |
| 12 |  | 75 | 93.6 | [44] |
| 13 |  | 83 | 93 | [88] |
| 14 |  | 90 | nm | [41] |
| 15 |  | 72 | nm | [16] |
| 16 |  | 91 | 90 (de.) | [34] |
| 17 |  | 90 | nm | [30] |
| 18 |  | 92 | >90 | [17] |
| 19 |  | 92 | 94 | [82],[68] |
| 20 |  | 92 | 91 | [18] |
| 21 |  | nm | 63 | [42] |
| 22 |  | 99 | 92 | [19] |
| 23 |  | 72 | 98 (de) | [29] |
| 24 |  | 93 | 97 | [26] |
| 25 |  | 70 | >95 | [20] |
| 26 |  | 75 | 99 | [37] |
| 27 |  | 85 | 91 | [82],[25],[50],[49] |
| 28 |  | 91 | >94 | [21] |
| 29 |  | 92 | >99 | [33] |
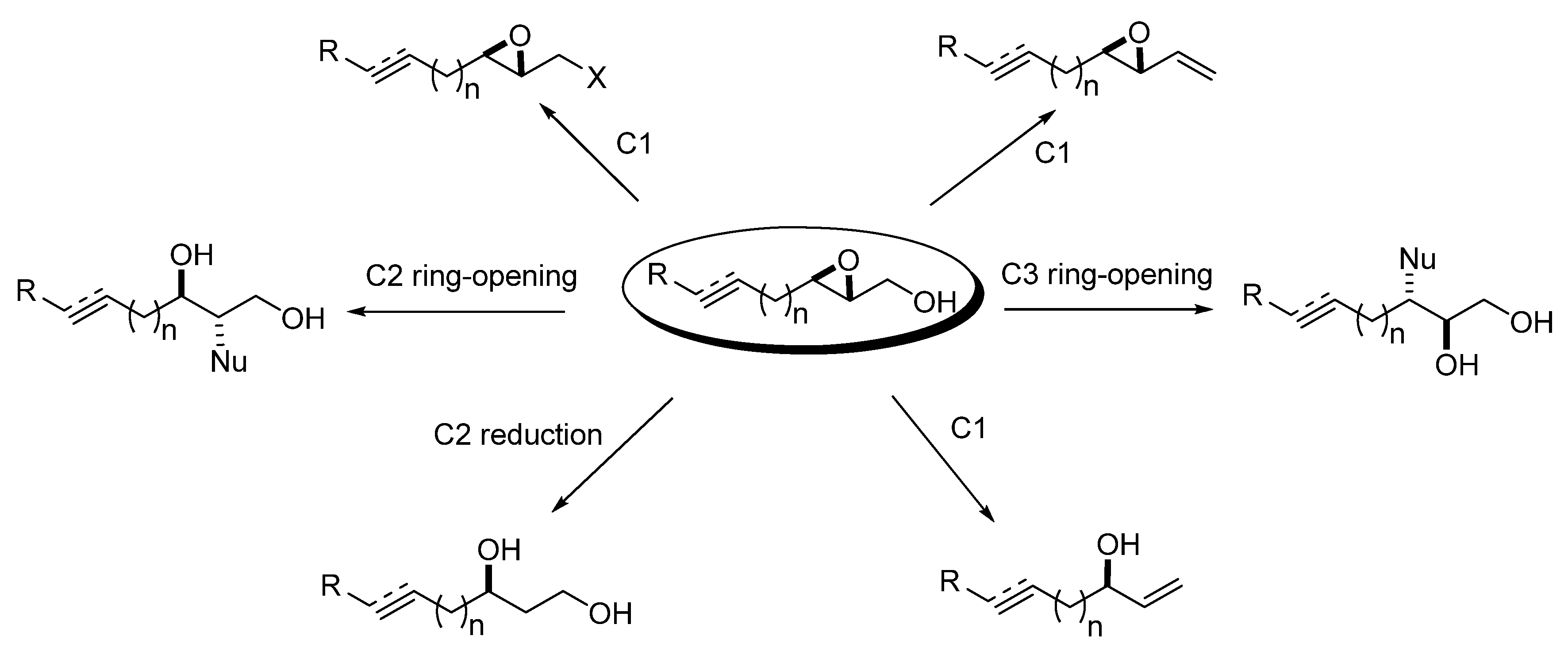
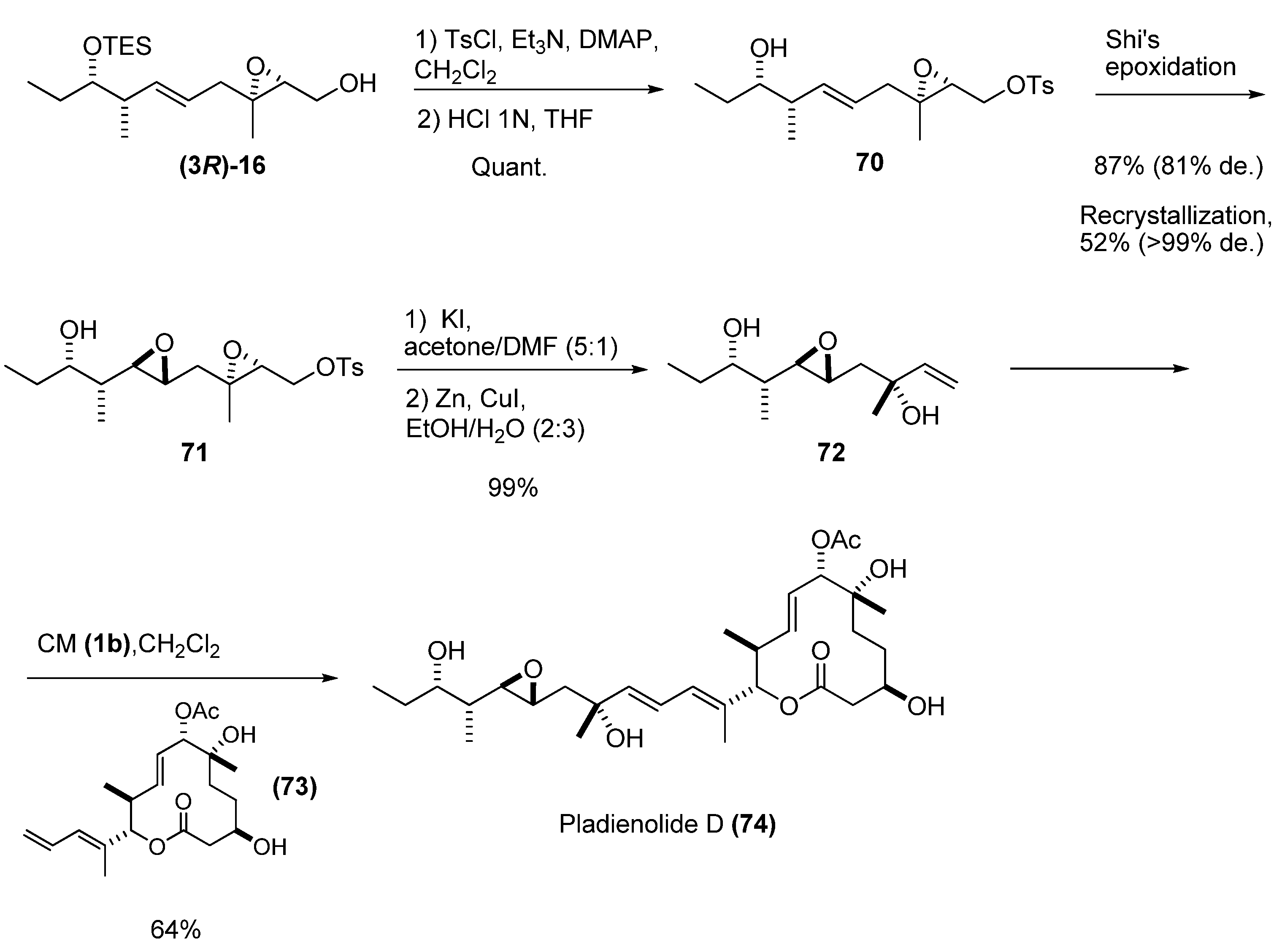
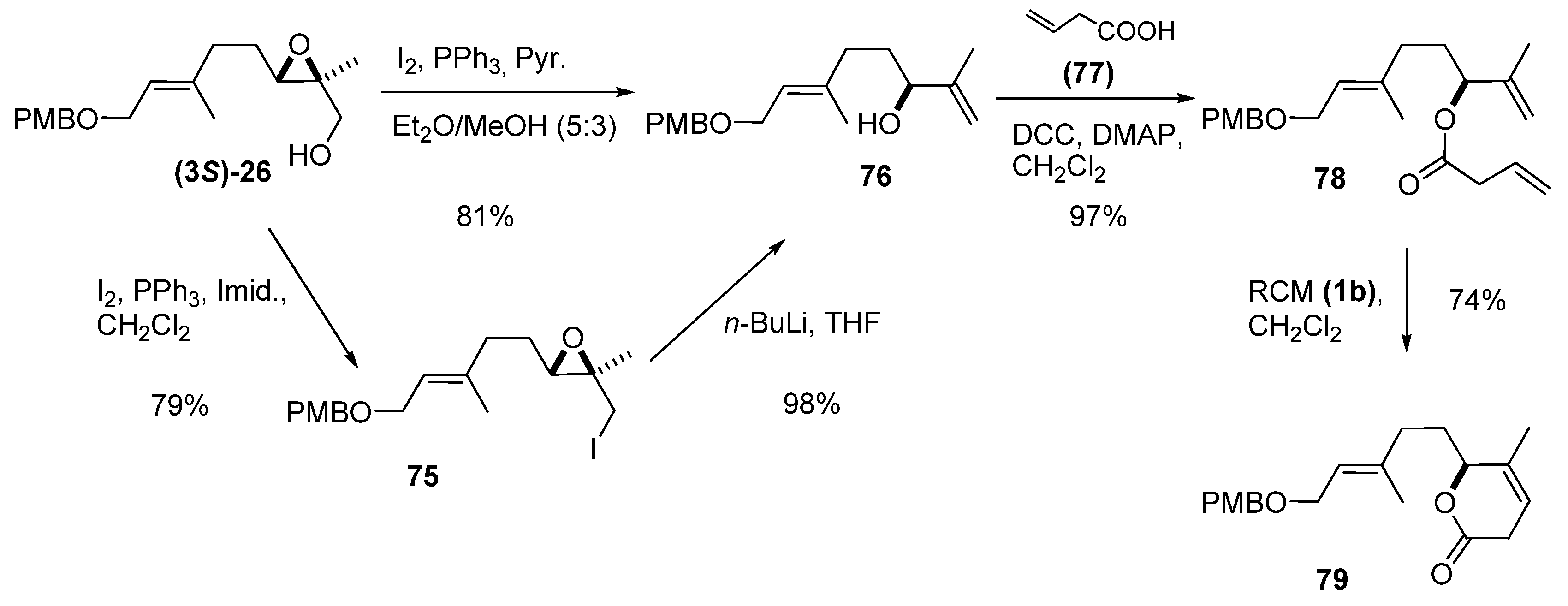
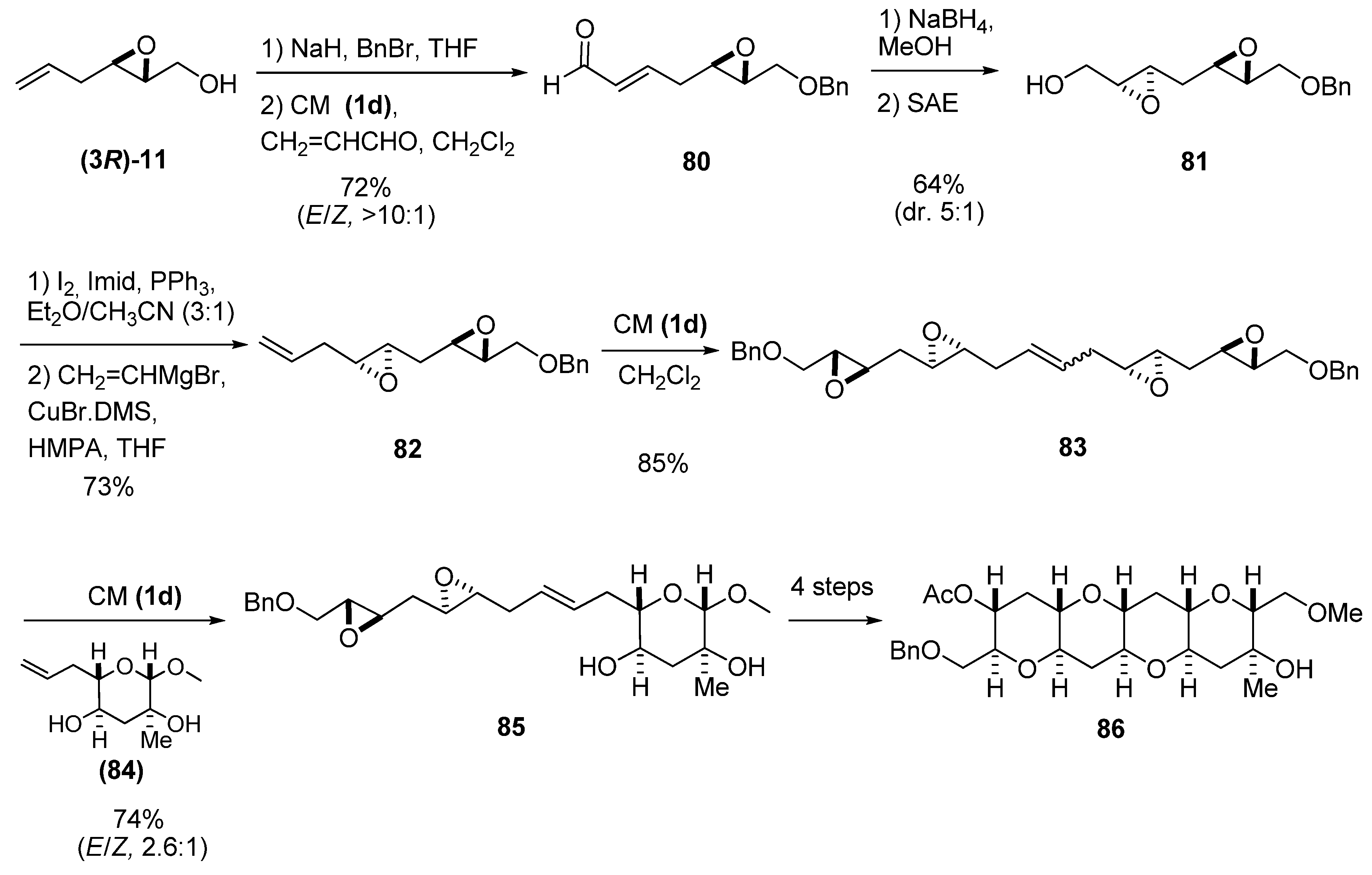
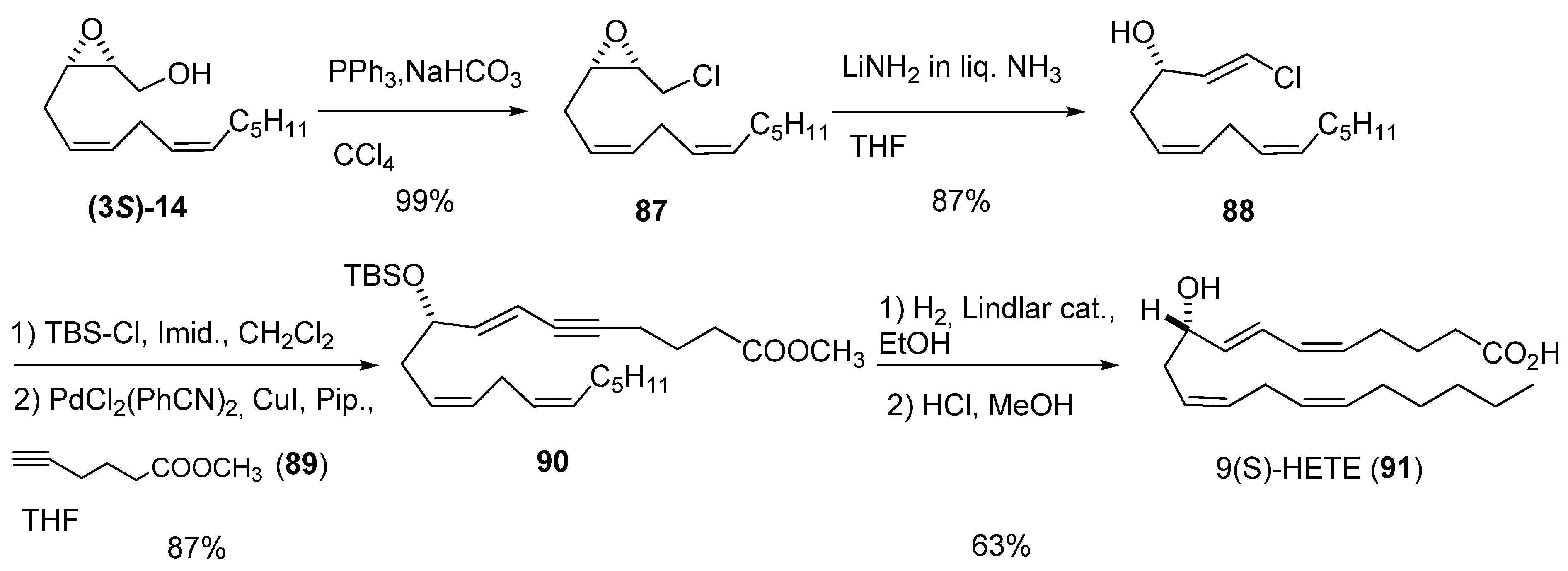
3. Transformations of C1













4. Epoxide Ring-Opening at C2
4.1. Nucleophilic attack at C2 with carbon nucleophiles



4.2. Nucleophilic reduction at C2 with hydride




4.3. Nucleophilic attack at C2 with nitrogen nucleophiles




5. Epoxide Ring-Opening at C3
5.1. Nucleophilic attack at C3 with carbon nucleophiles

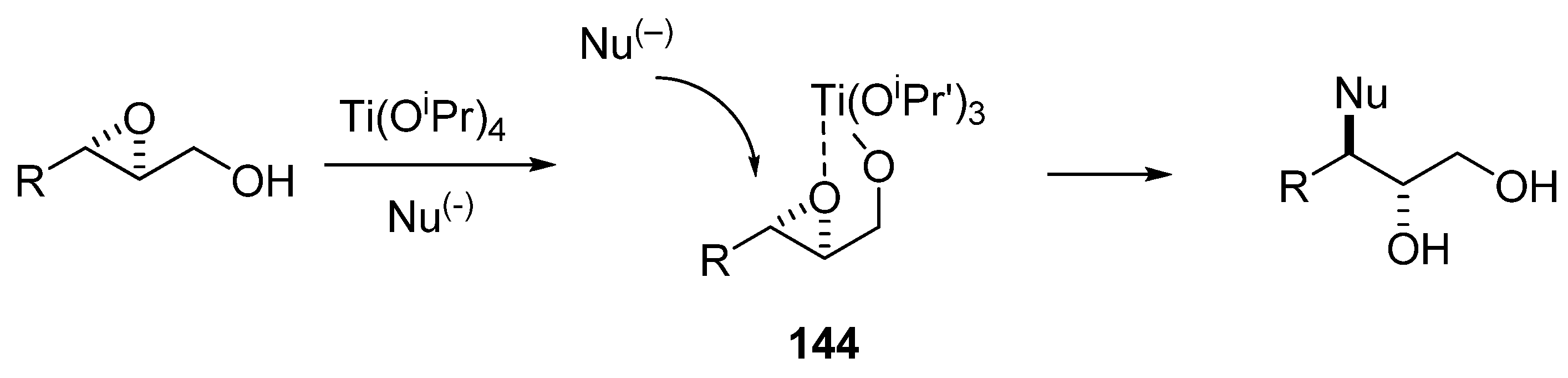
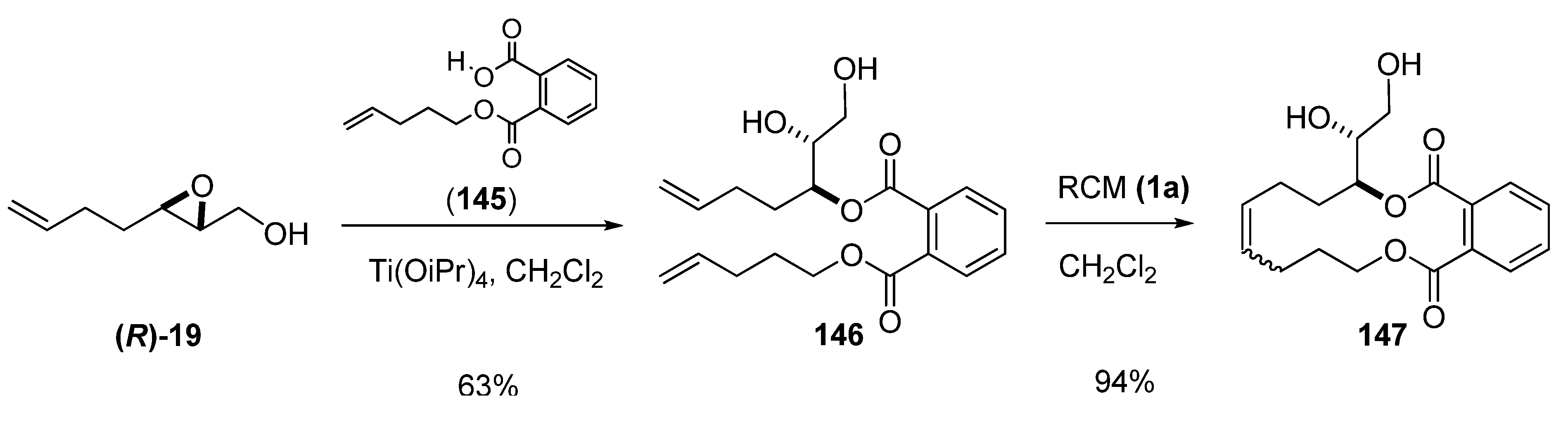
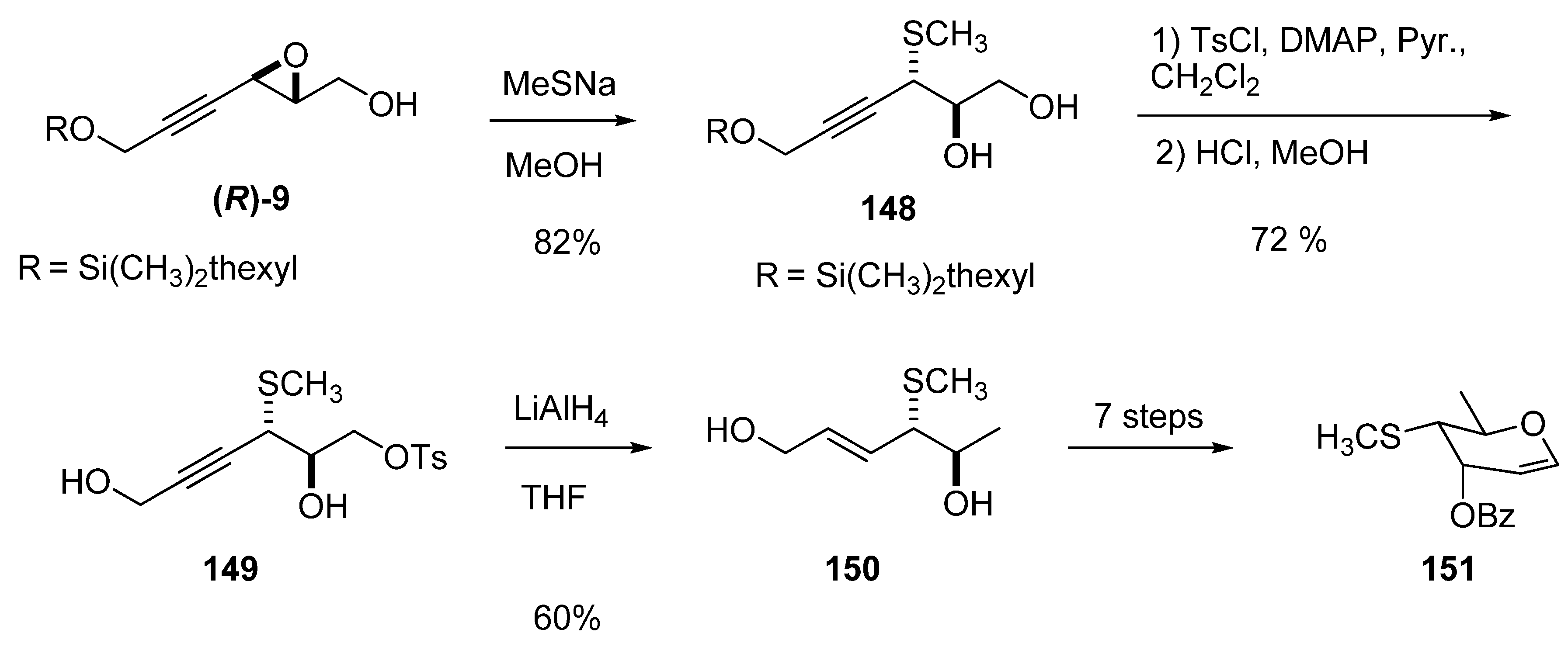
5.2. Nucleophilic attack at C3 with oxygen or sulfur nucleophiles



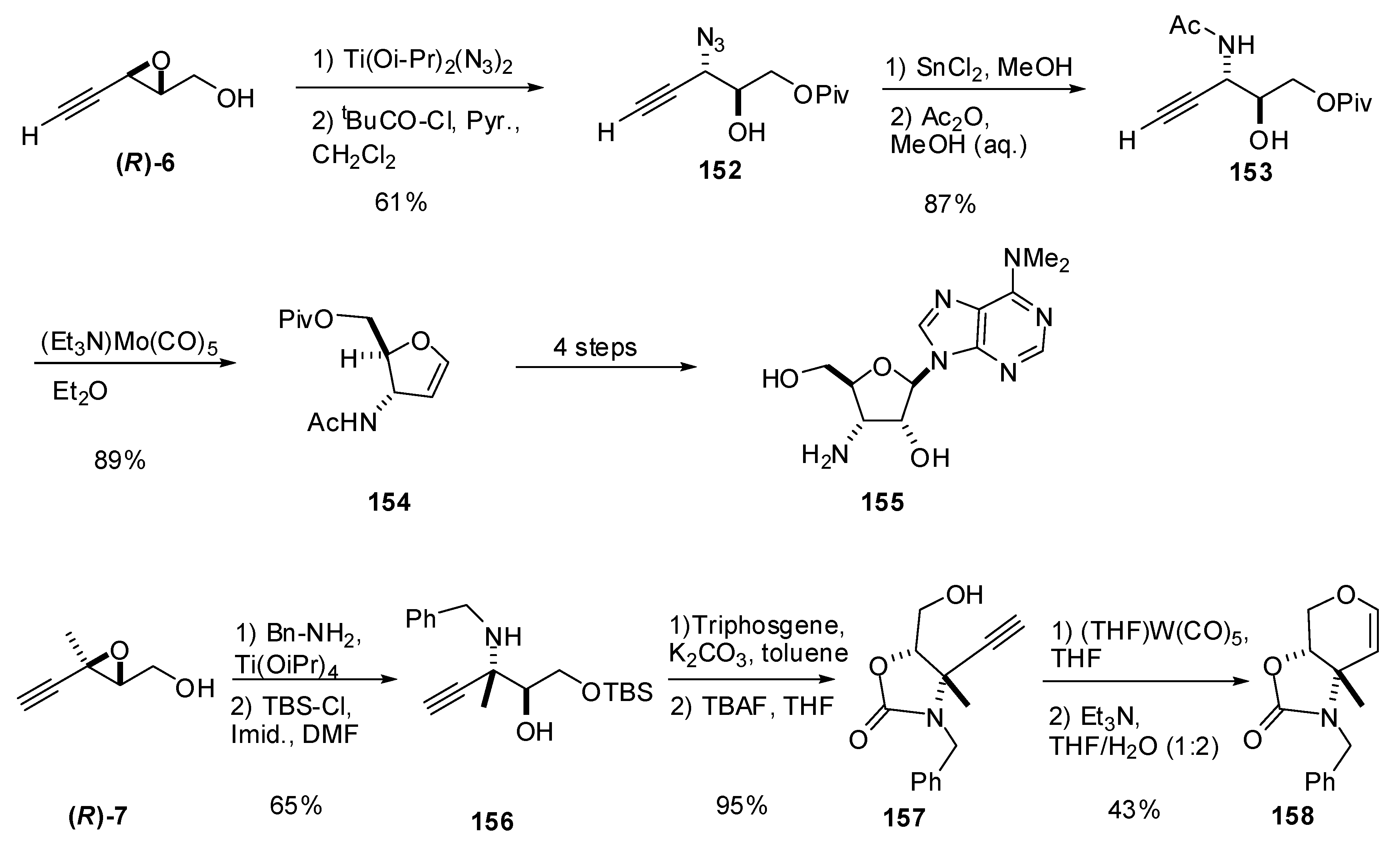
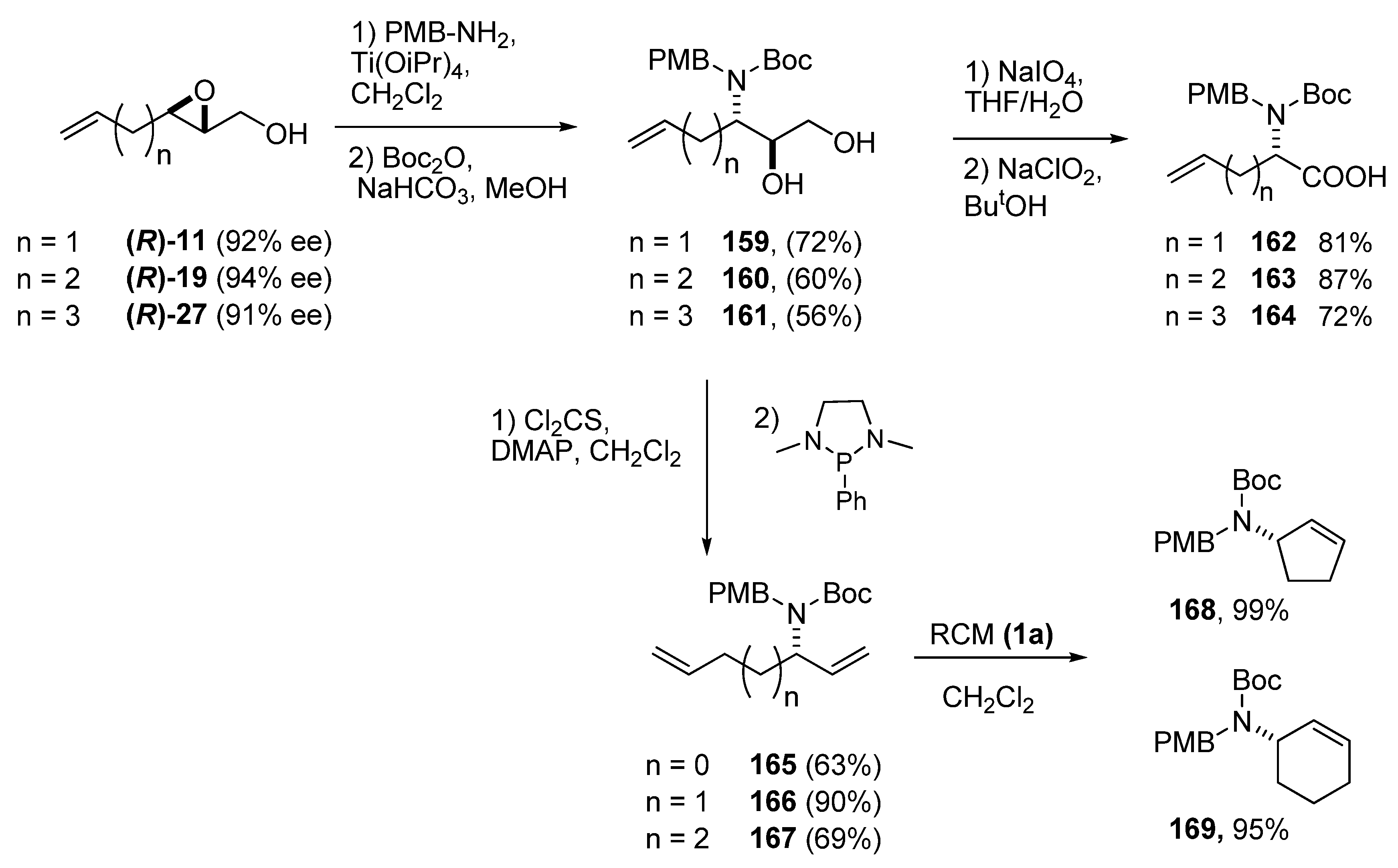
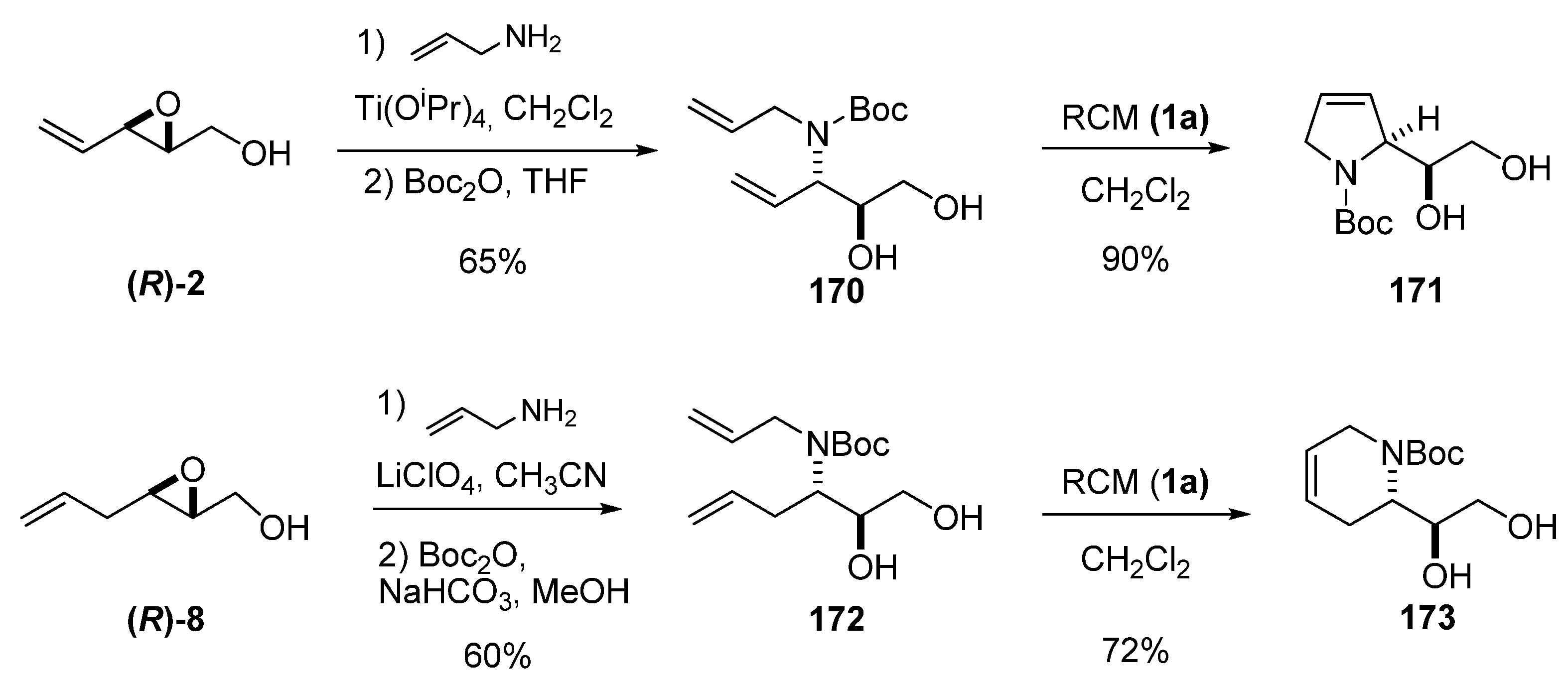
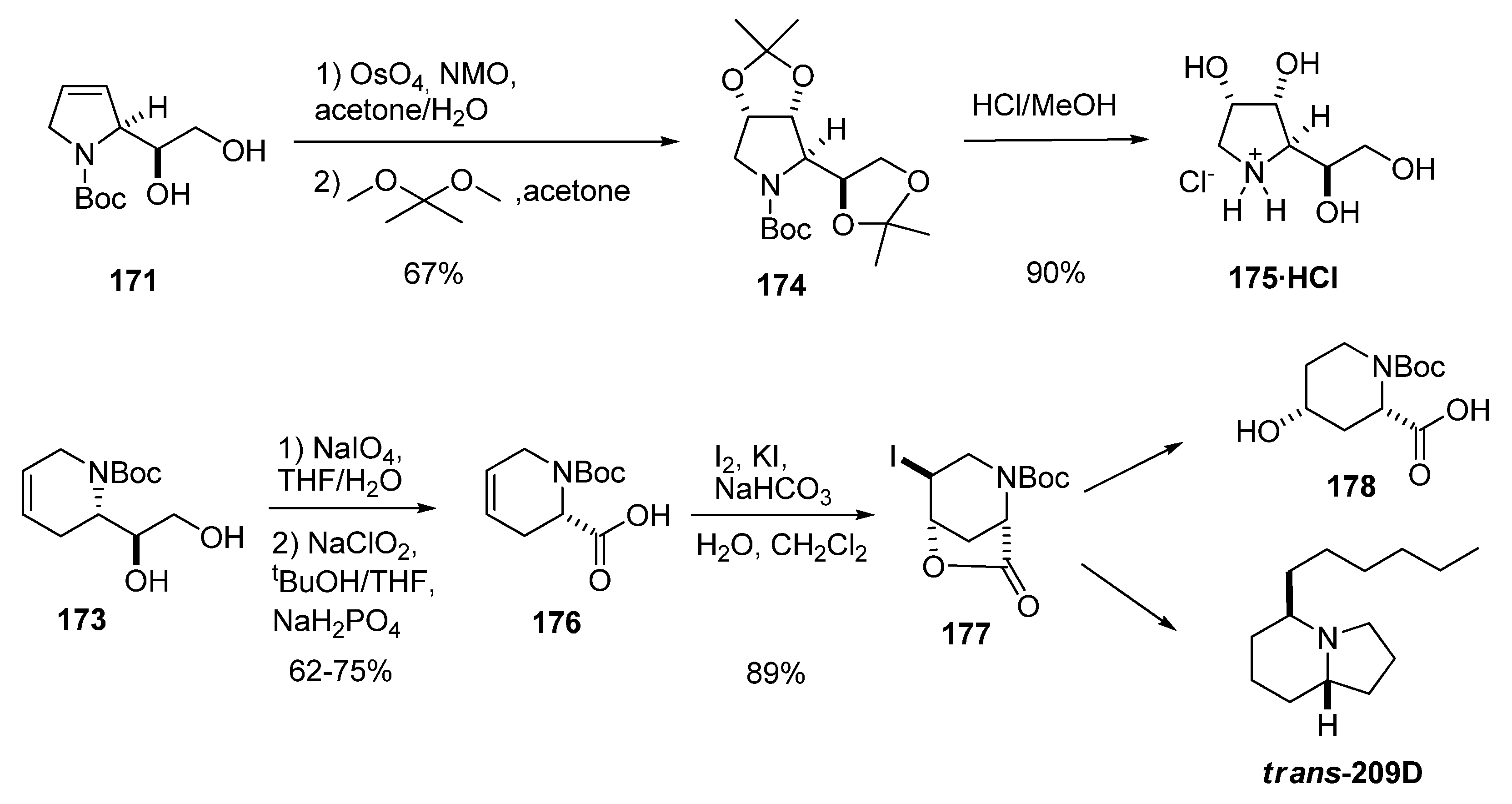
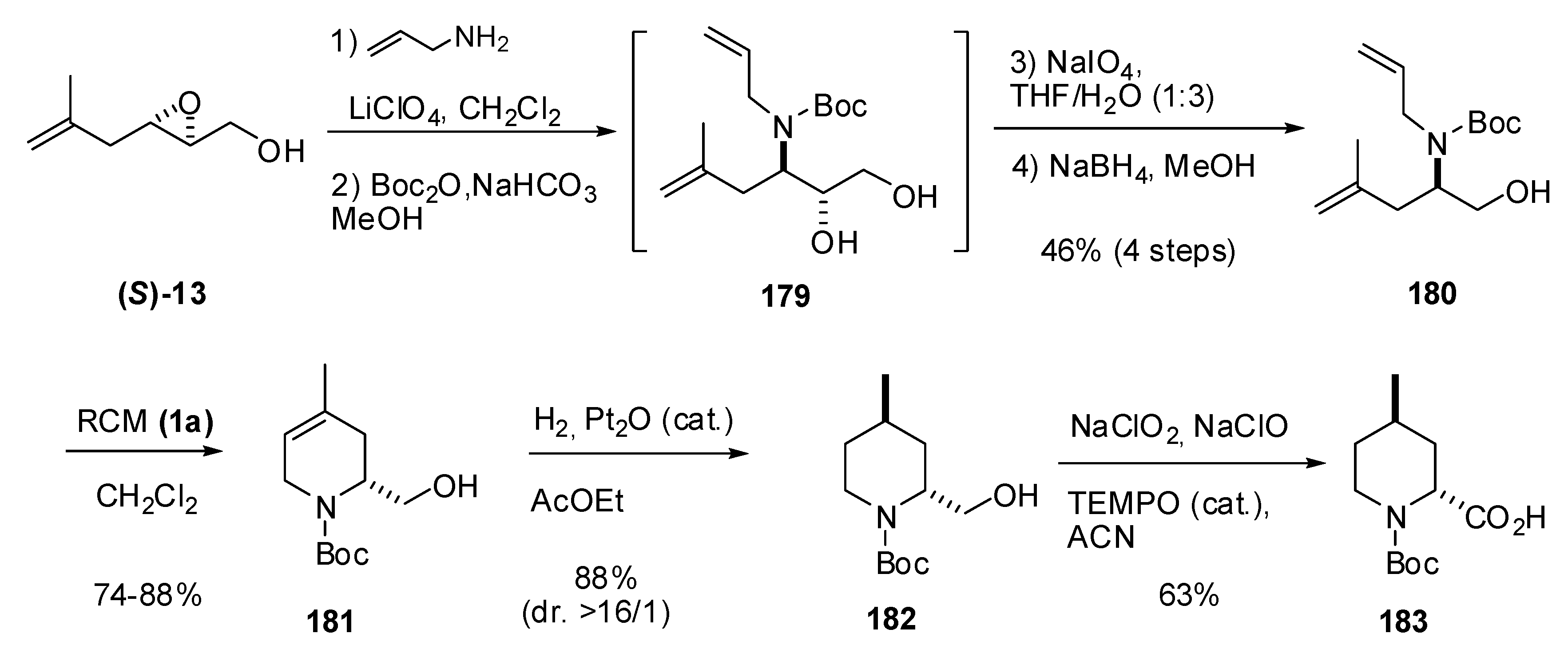
5.3. Nucleophilic attack at C3 with nitrogen nucleophiles




{kind=link}
{kind=link}
{kind=link}
{kind=link}
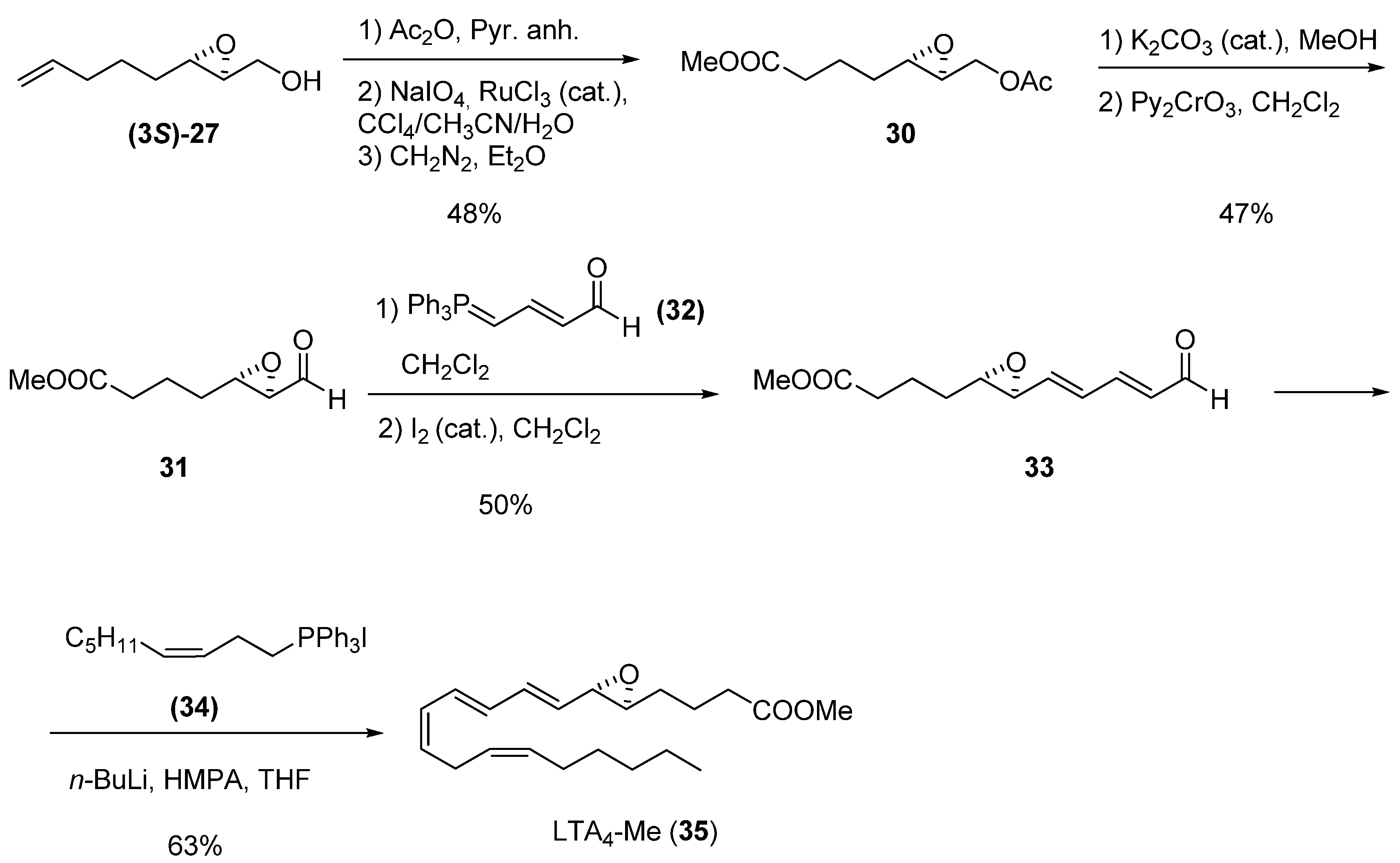{kind=link}
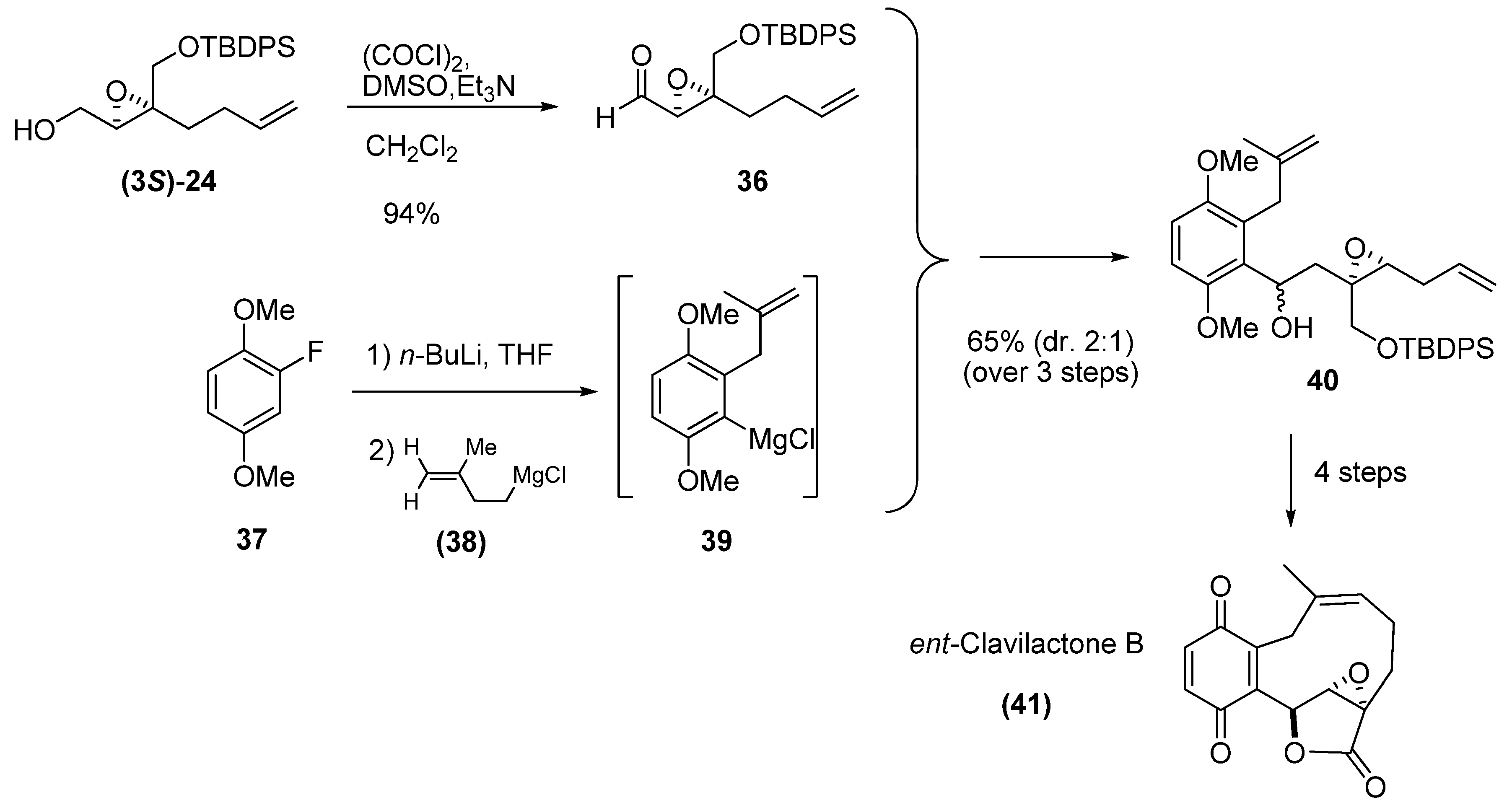{kind=link}
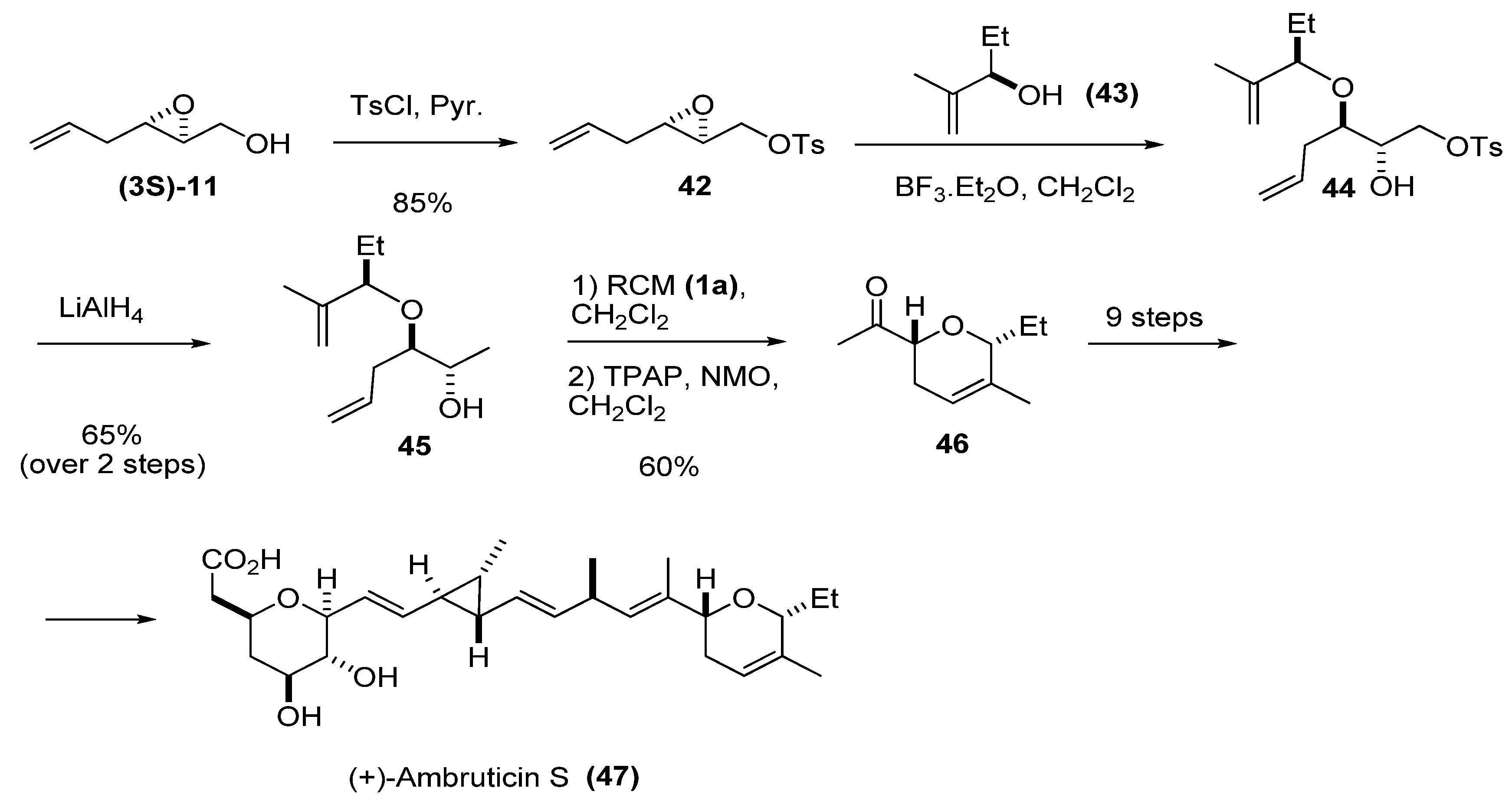{kind=link}
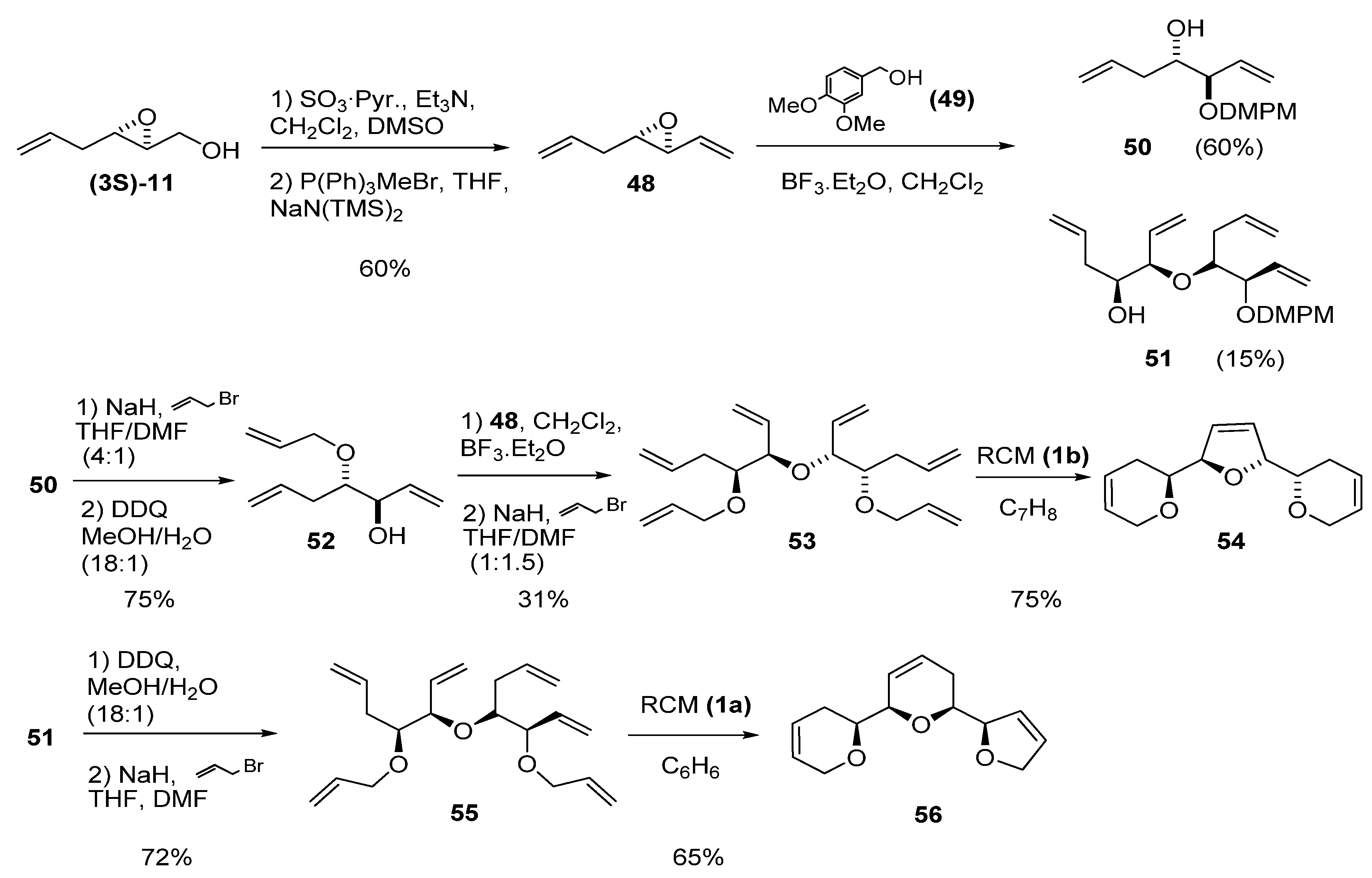{kind=link}
{kind=link}
{kind=link}
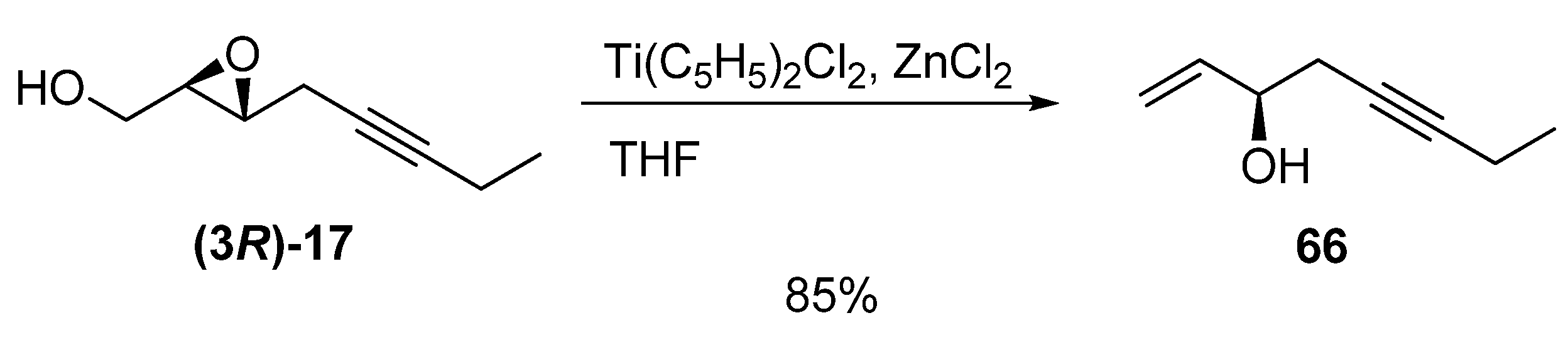{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
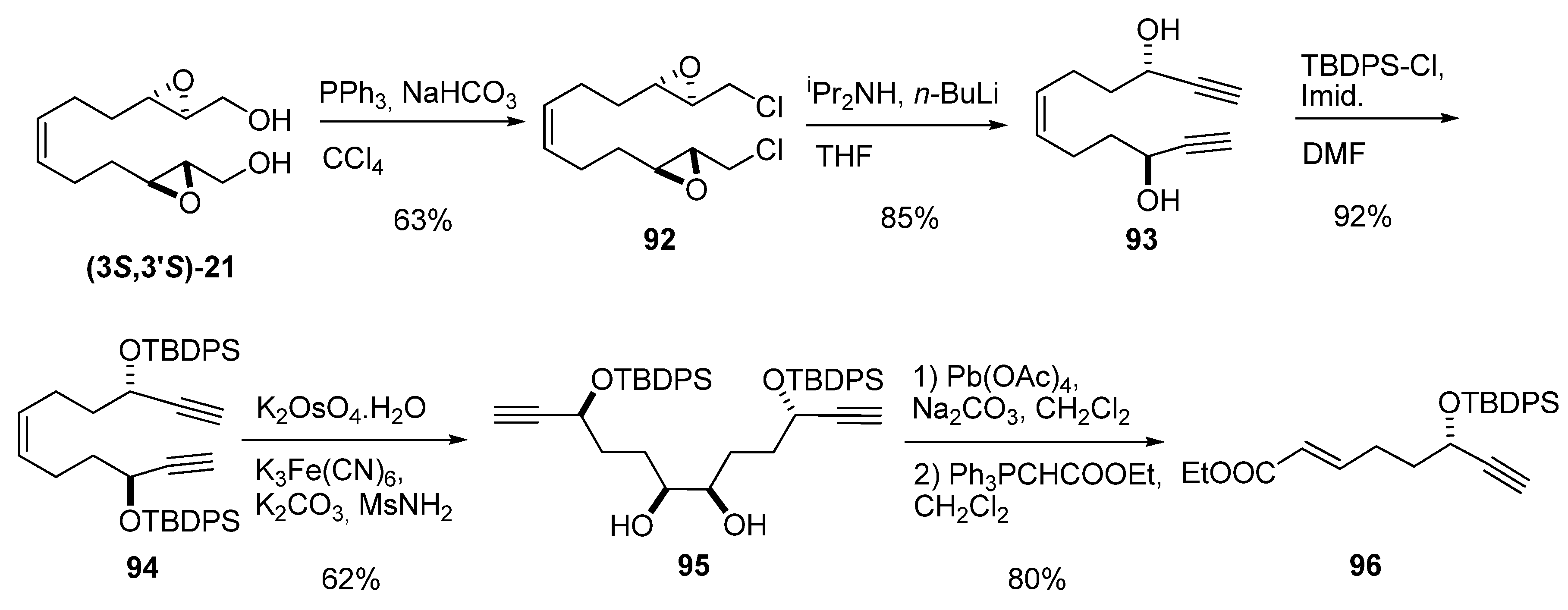{kind=link}
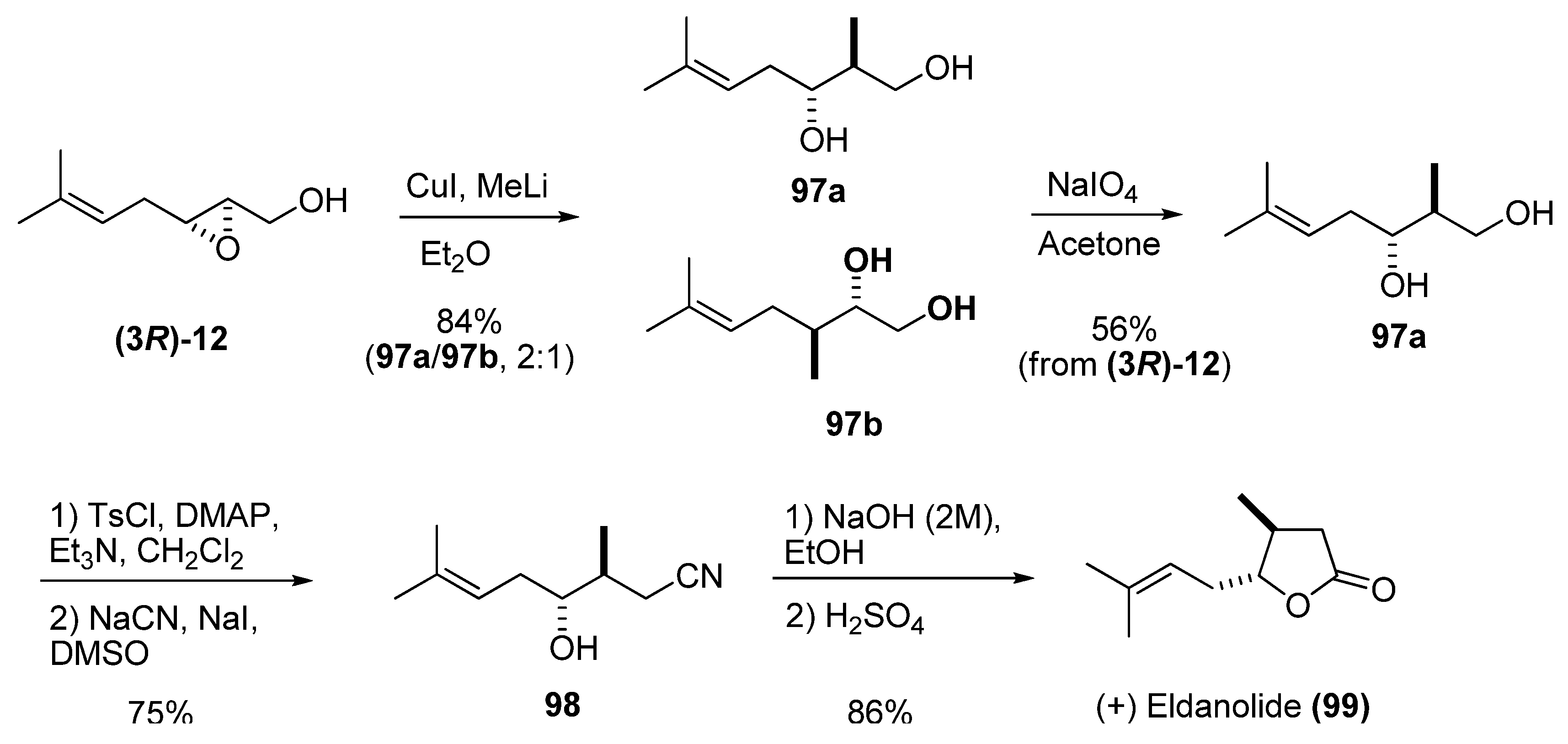{kind=link}
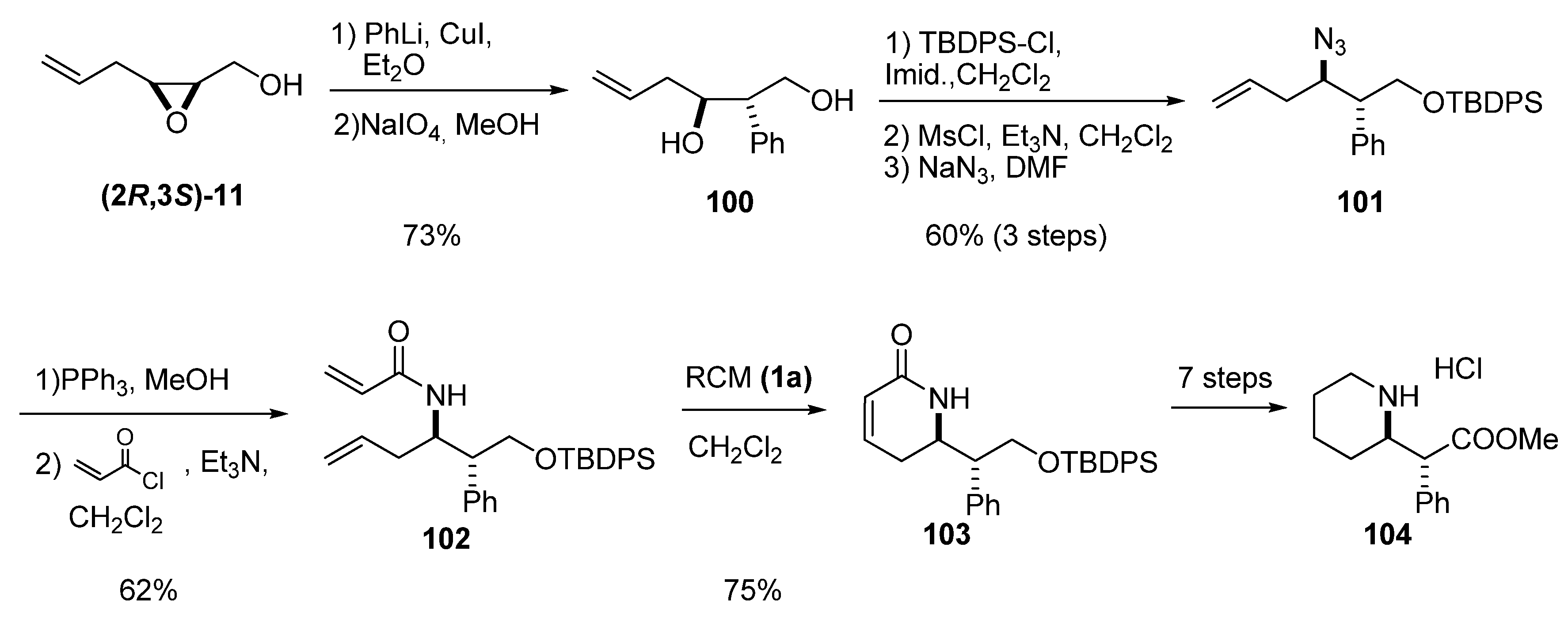{kind=link}
{kind=link}
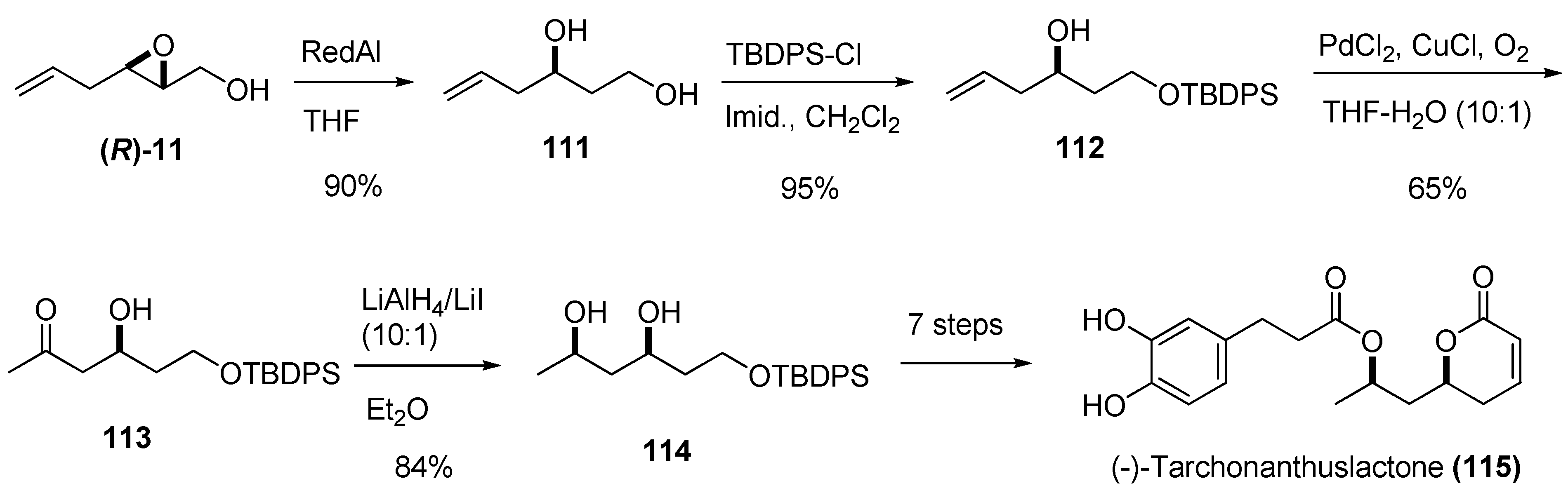{kind=link}
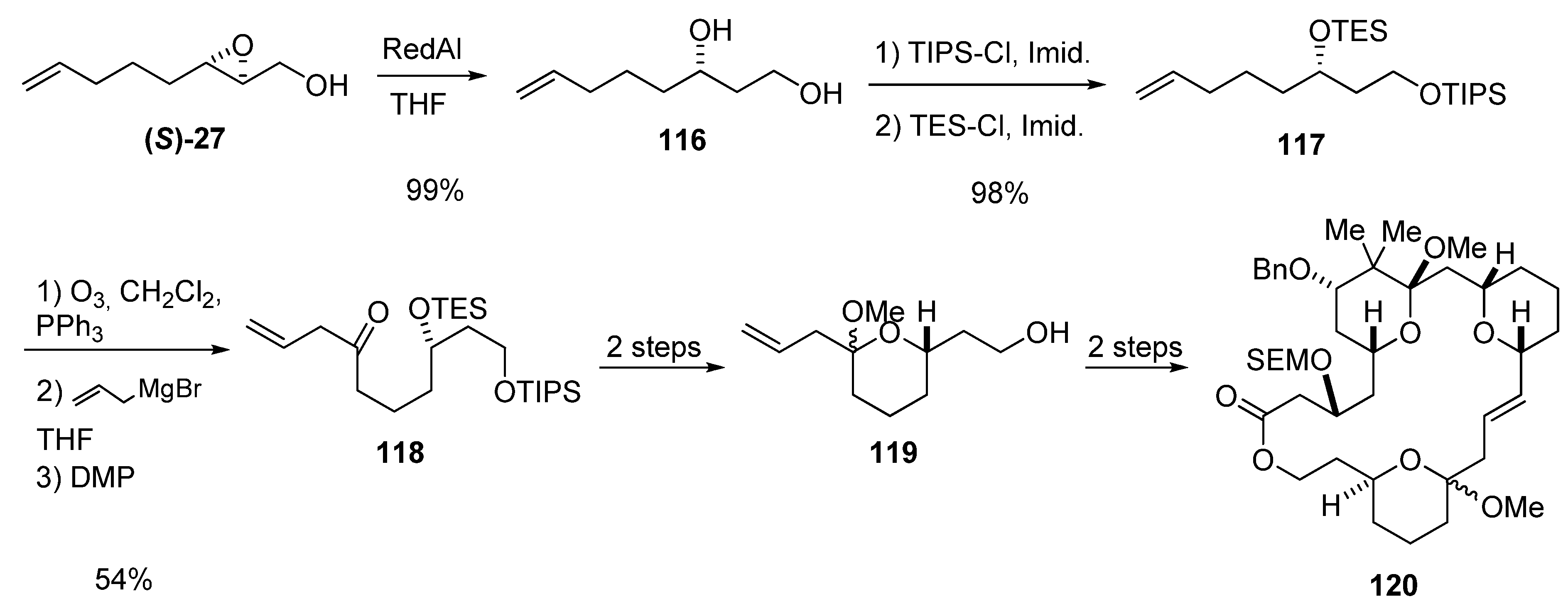{kind=link}
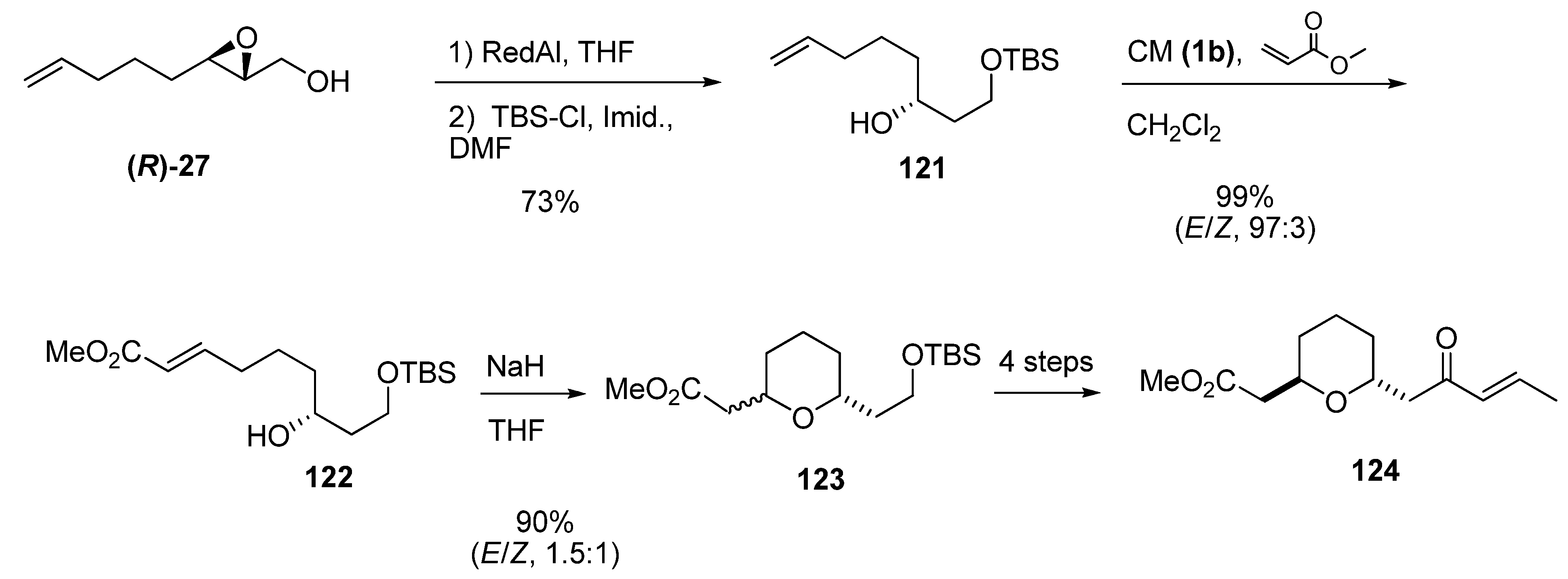{kind=link}
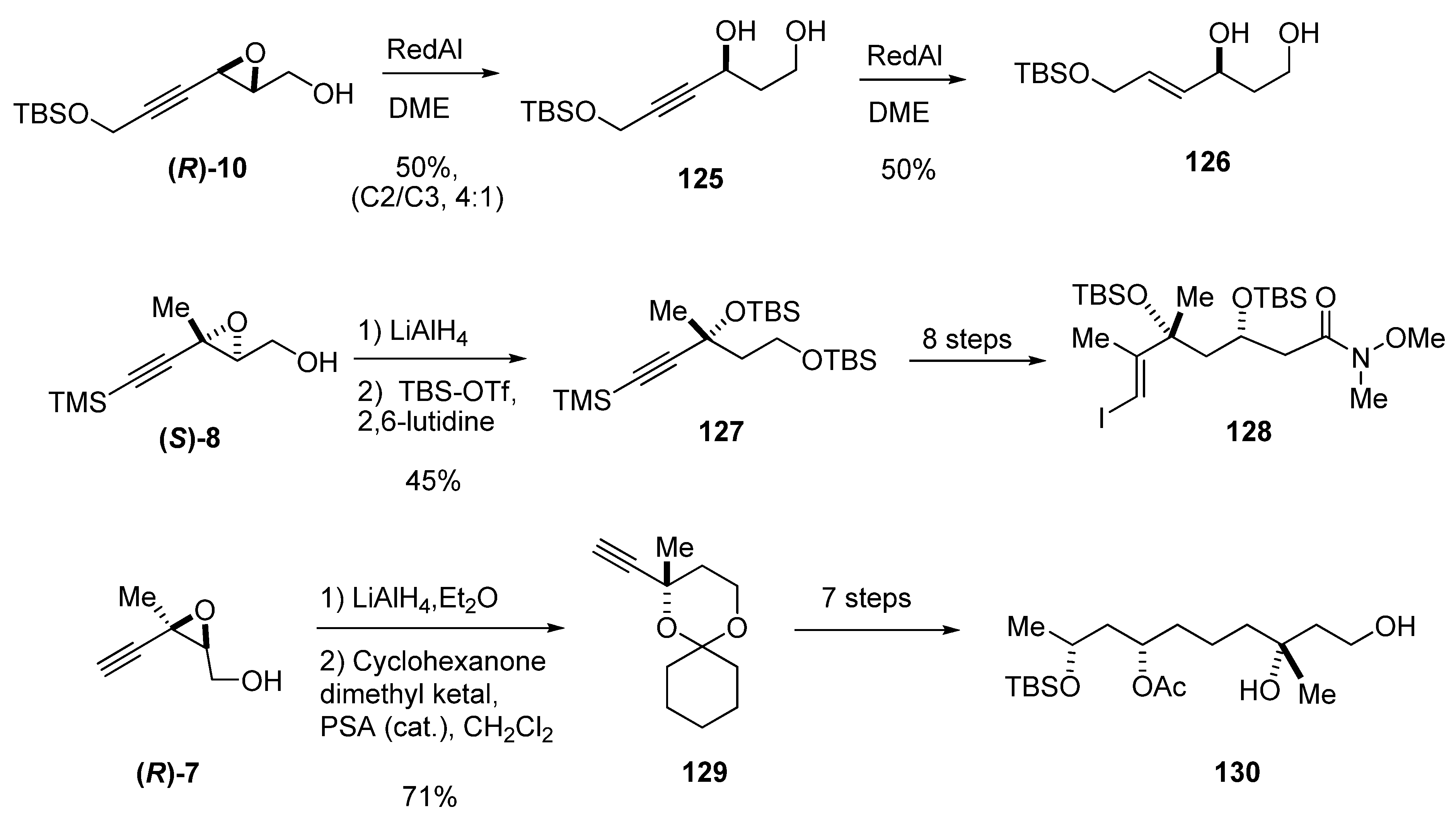{kind=link}
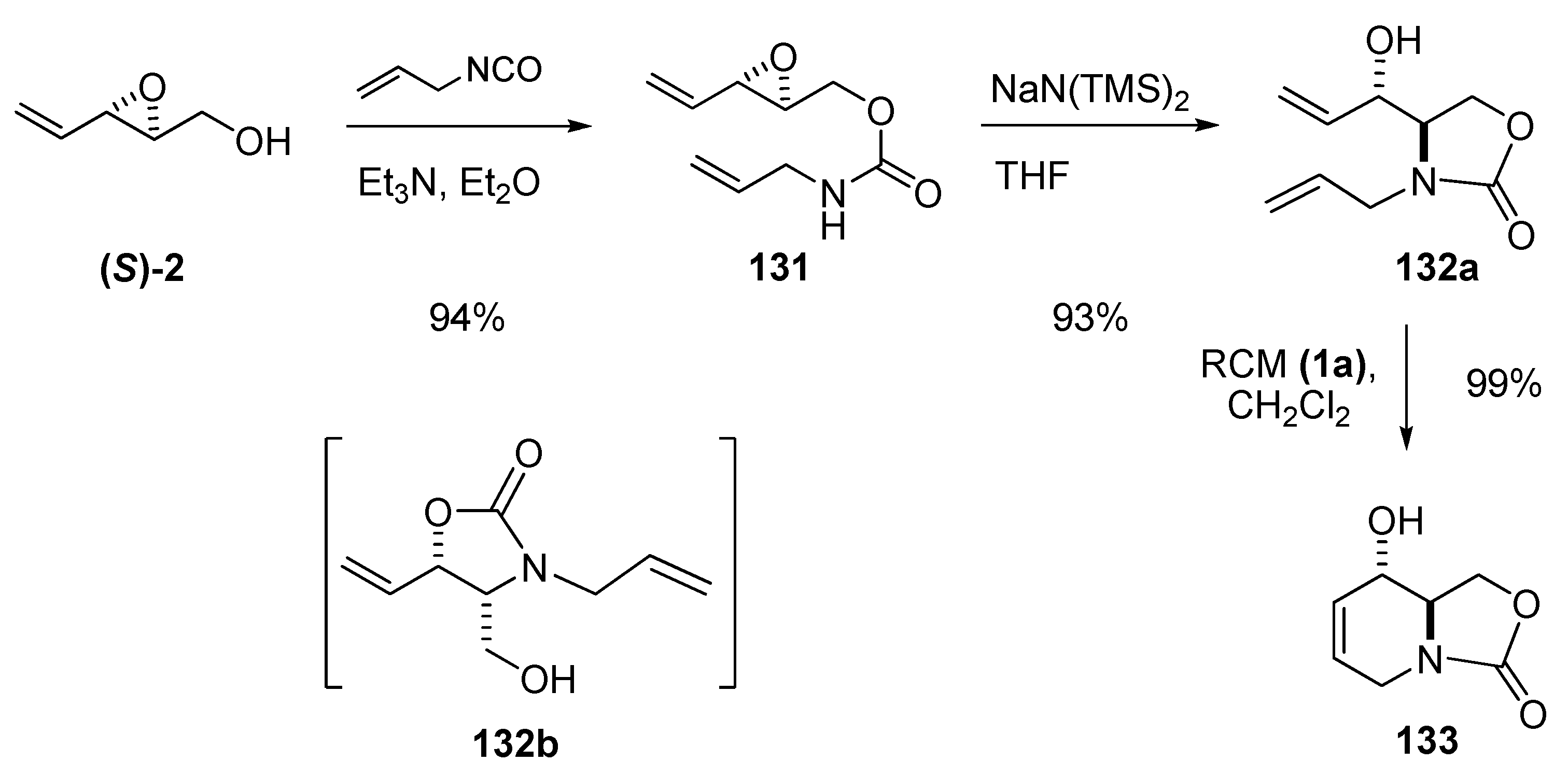{kind=link}
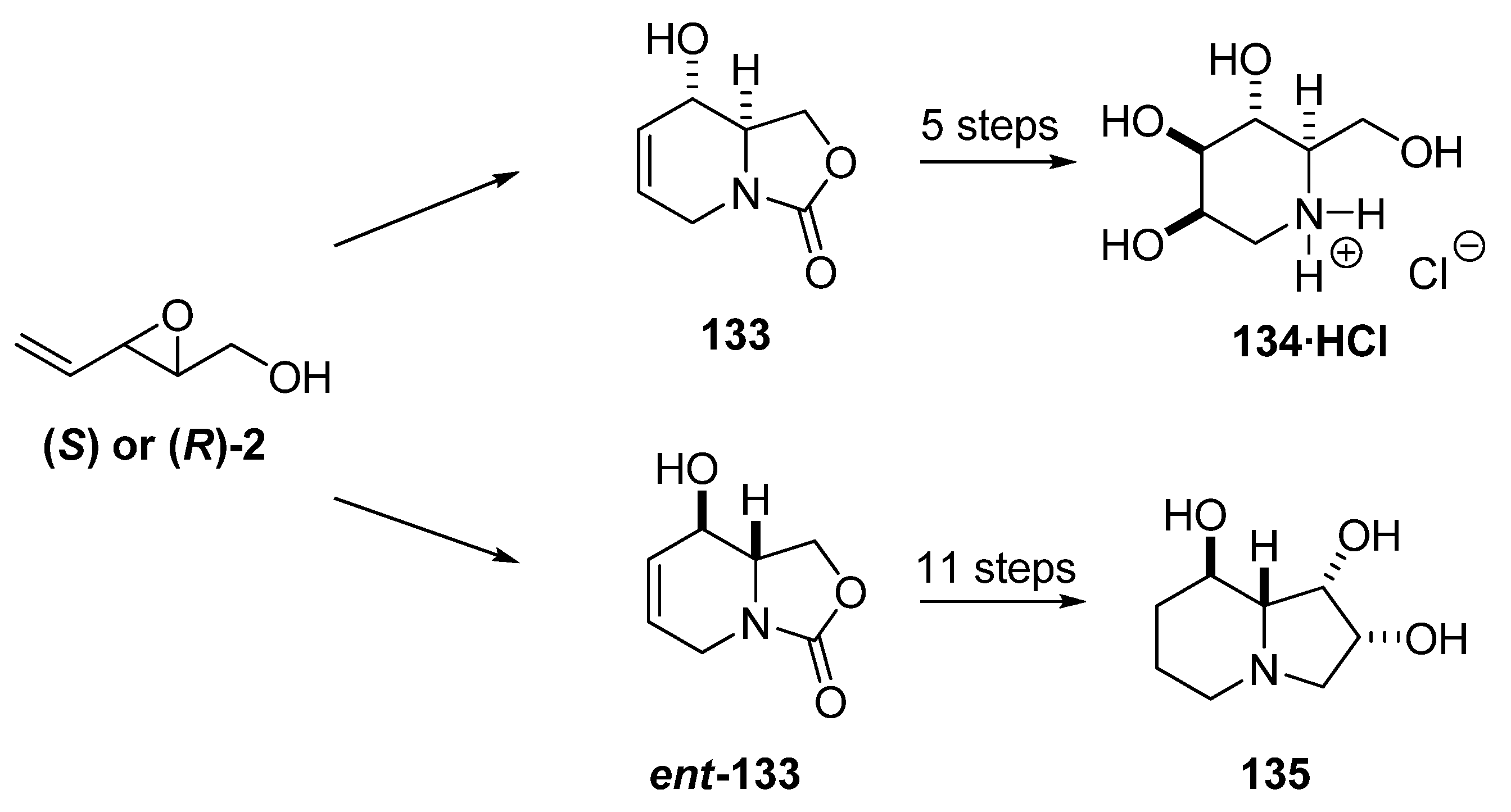{kind=link}
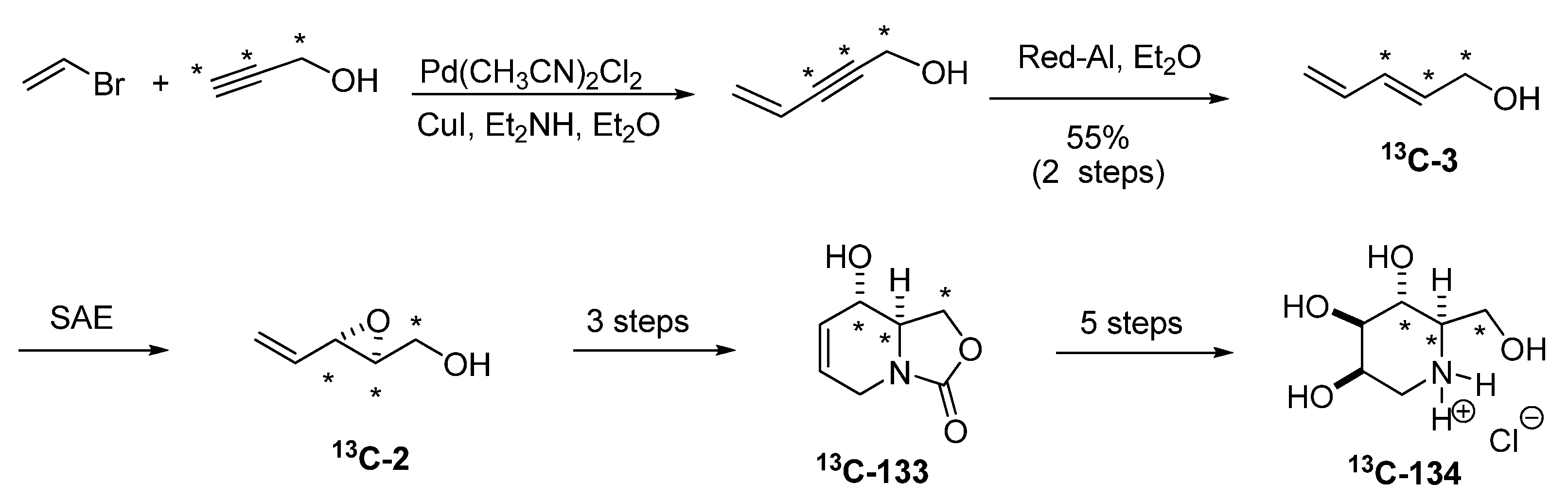{kind=link}
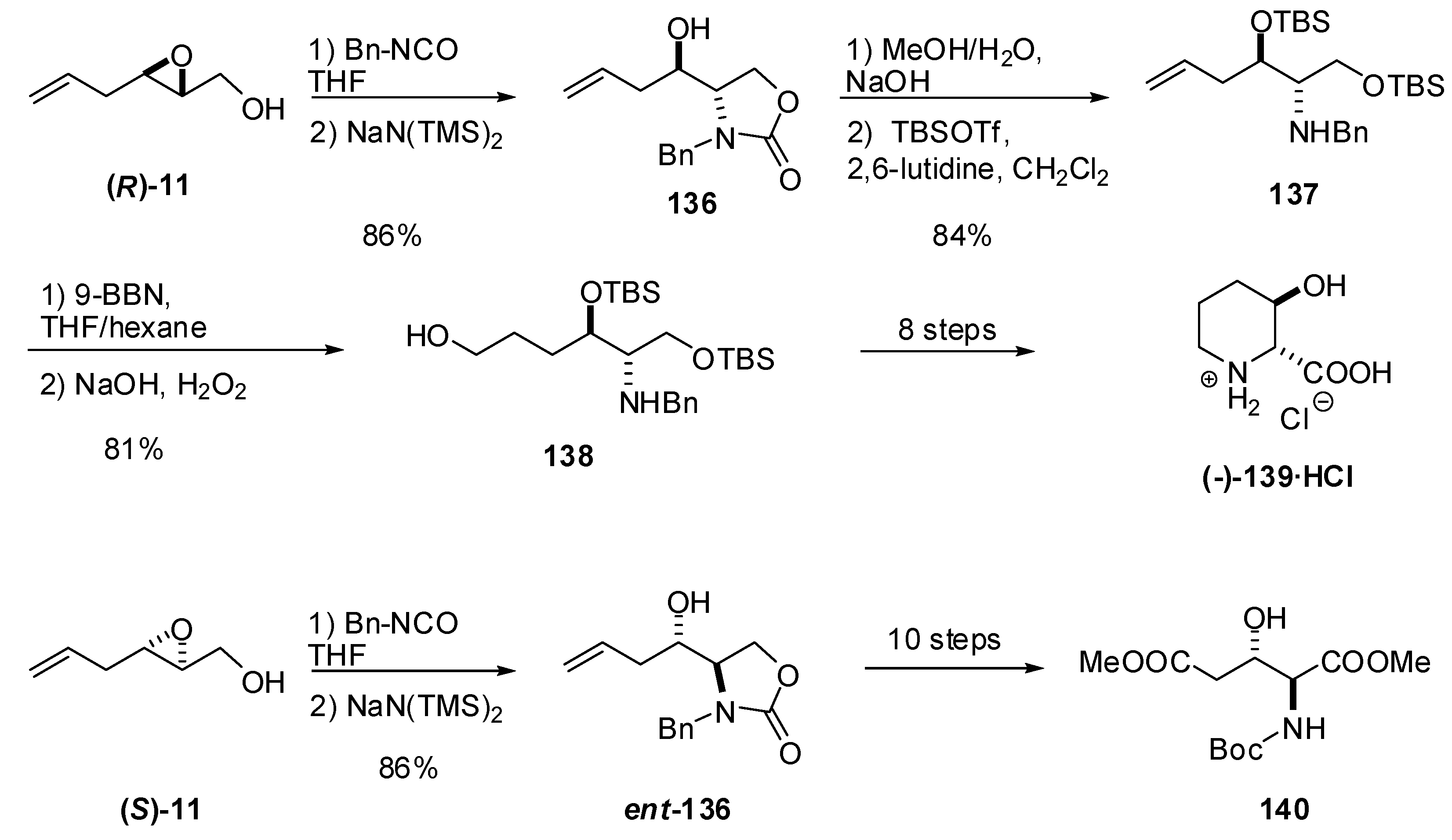{kind=link}
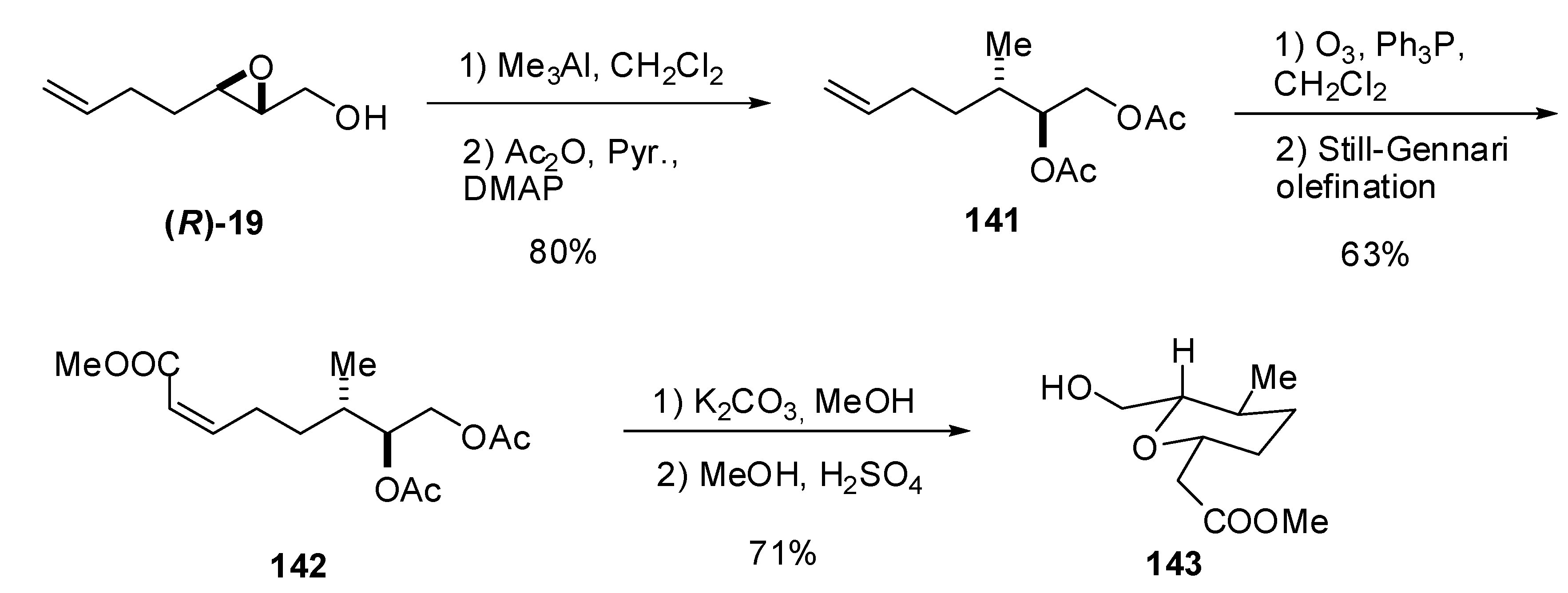{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
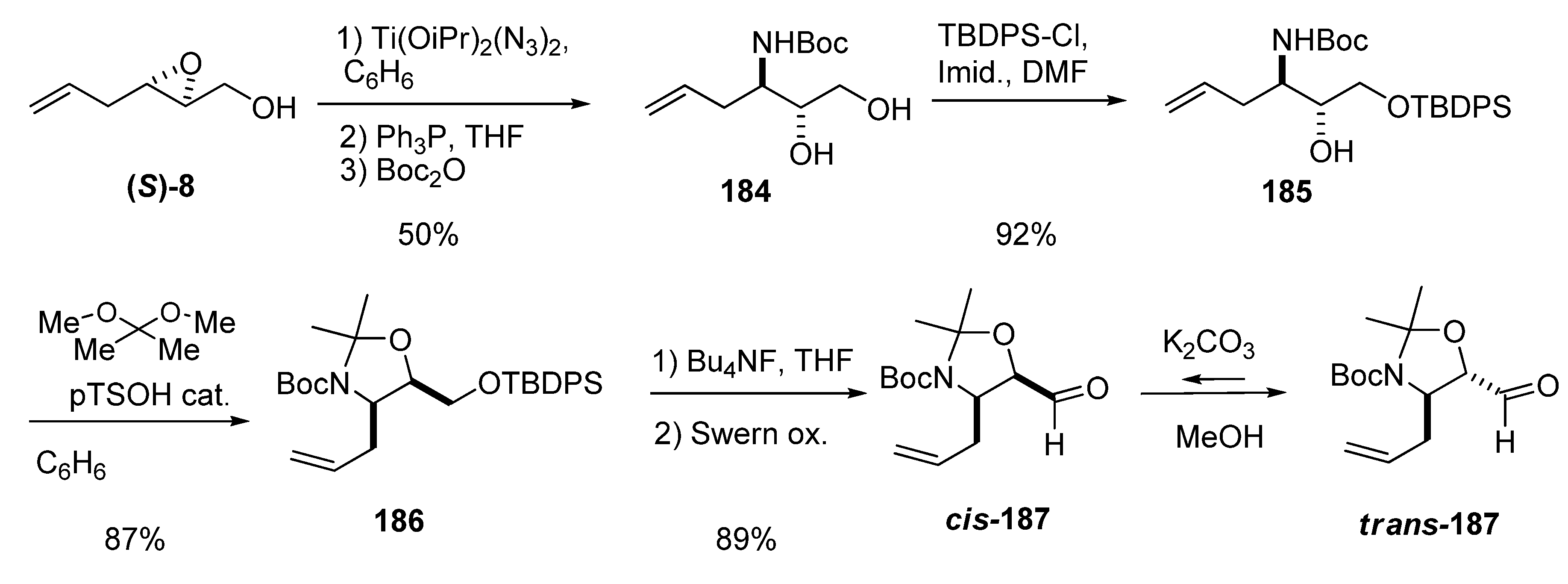{kind=link}
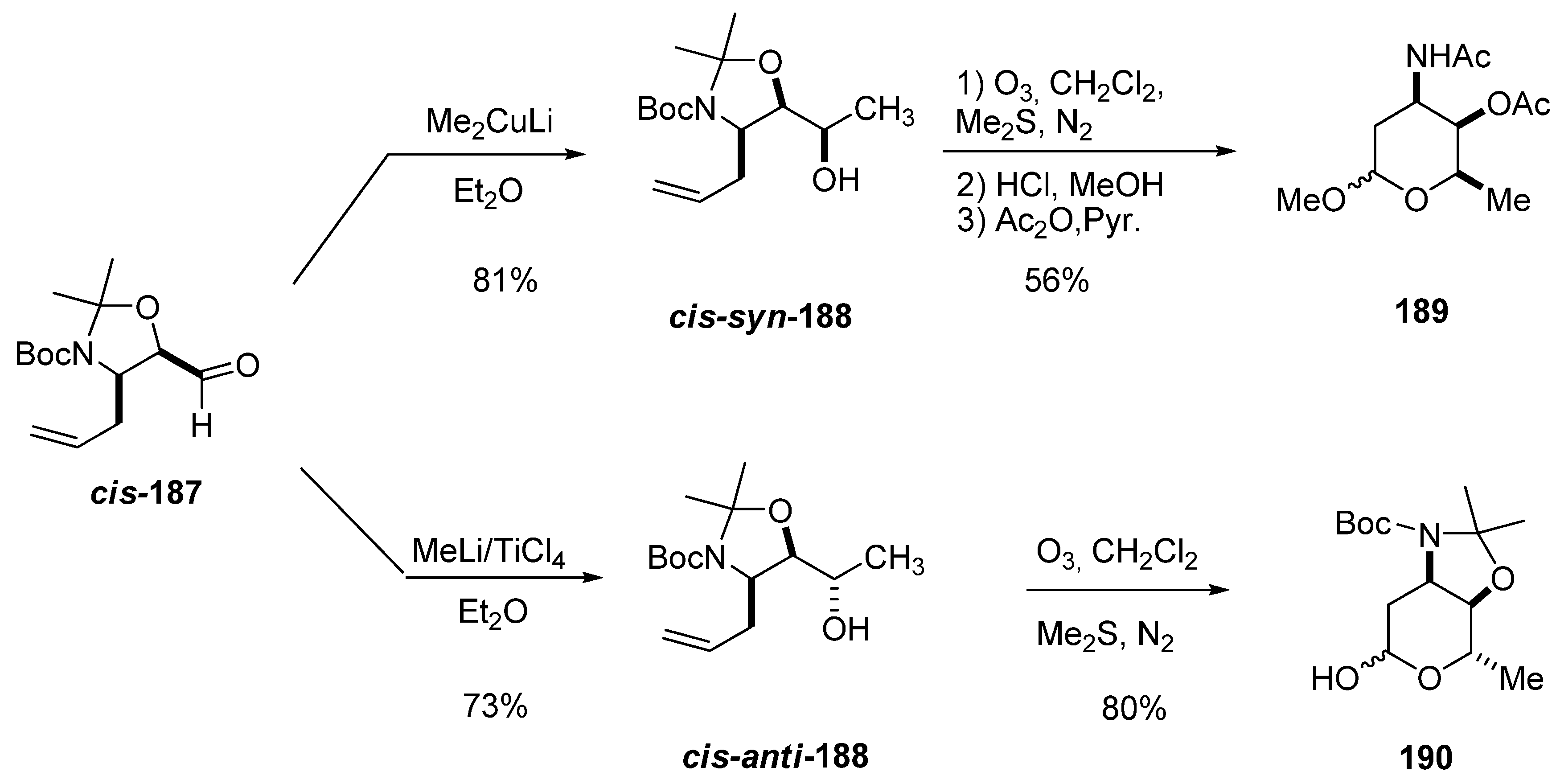{kind=link}
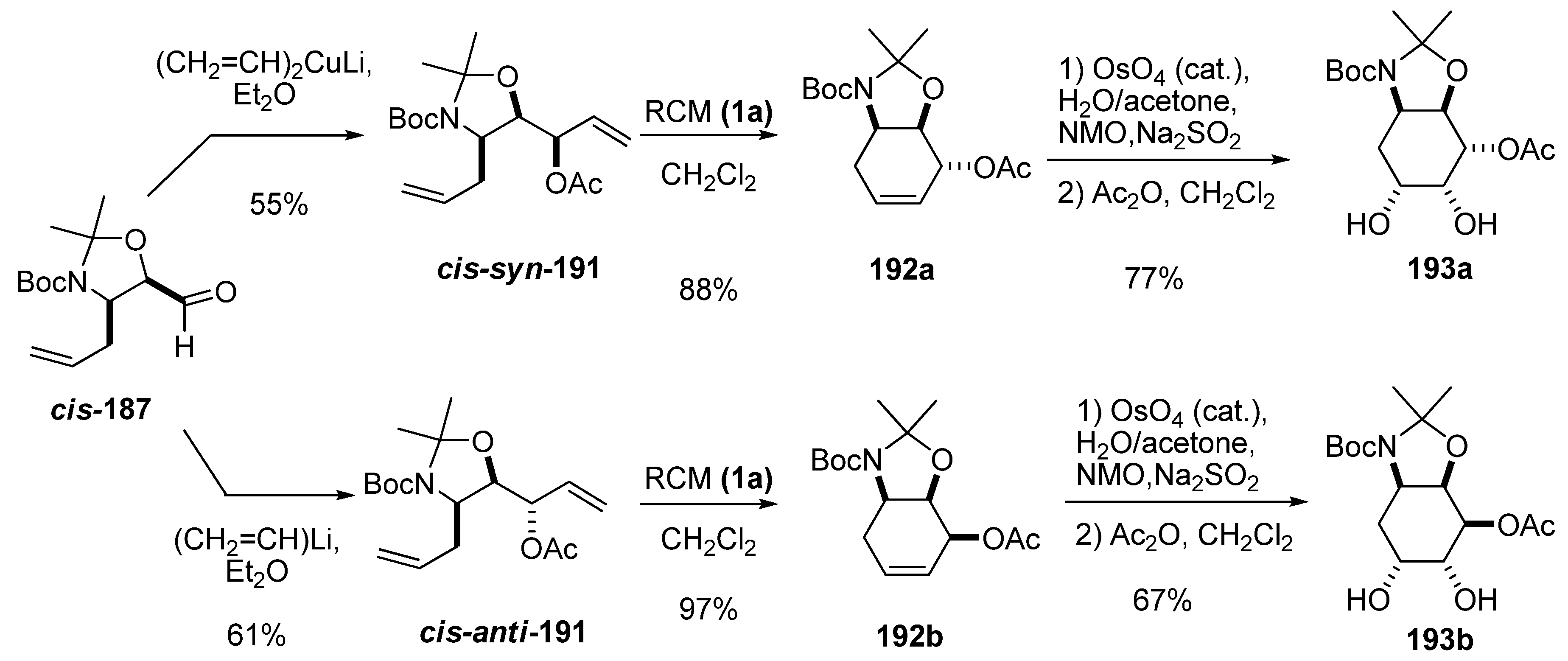{kind=link}




6. Conclusions
Acknowledgements
References and Notes
- Gao, Y.; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- Schinzer, D. Asymmetric synthesis.The Sharpless epoxidation. In Organic Synthesis Highlights II; VCH: Weinheim, Germany, 1995; pp. 3–8. [Google Scholar]
- Katsuki, T.; Martin, V. Asymmetric epoxidation of allylic alcohols: The Katsuki-Sharpless epoxidation reaction. Org. React. 1996, 48, 1–299. [Google Scholar]
- Grubbs, R.H.; Chang, S. Recent advances in olefin metathesis and its application in organic synthesis. Tetrahedron 1998, 54, 4413–4450. [Google Scholar] [CrossRef]
- Yet, L. Metal-mediated synthesis of medium-sized rings. Chem. Rev. 2000, 100, 2963–3007. [Google Scholar] [CrossRef]
- Van Otterlo, W.A.L.; de Koning, C.B. Metathesis in the synthesis of aromatic compounds. Chem. Rev. 2009, 109, 3743–3782. [Google Scholar]
- Grubbs, R.H.; Miller, S.J.; Fu, G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995, 28, 446–452. [Google Scholar] [CrossRef]
- Gibson, S.E.; Keen, S.P. Cross-metathesis. In Topics in Organometallic Chemistry; Springer-Verlag: Berlin, Germany, 1998; pp. 155–181. [Google Scholar]
- Connon, S. Recent developments in olefin cross-metathesis. Angew. Chem. Int. Ed. 2003, 42, 1900. [Google Scholar] [CrossRef]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J.; Hoveyda, A.H. A recyclable Ru-based metathesis catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar]
- Jaeger, V.; Huemmer, W.; Stahl, U.; Gracza, T. Controlled synthesis of enantio-, regio-, and diastereomers of amino-4-pentenediols from 1,4-pentadien-3-ol via epoxy-4-pentenols I. Erythro-1-amino-4-pentene-2,3-diols. Synthesis 1991, 769–776. [Google Scholar]
- Jaeger, V.; Schroeter, D.; Koppenhoefer, B. Asymmetric sharpless epoxidation of divinylcarbinol. erythro-D and -L-4-pentenitols by hydrolysis of regioisomeric epoxy-4-pentenols. Tetrahedron 1991, 47, 2195–2210. [Google Scholar] [CrossRef]
- Jaeger, V.; Stahl, U.; Huemmer, W. Controlled synthesis of regio-, enantio-, and diastereomers of amino-4-pentenediols from 1,4-pentadien-3-ol via epoxy-4-pentenols. II. erythro- and threo-3-amino-4-pentene-1,2-diols and erythro-2-benzylamino-4-pentene-1,3-diol. Synthesis 1991, 776–782. [Google Scholar]
- Payne, G.B. Epoxide migrations with alpha, beta-epoxy alcohols. J. Org. Chem. 1962, 27, 3819–3822. [Google Scholar] [CrossRef]
- Taber, D.F.; Zhang, Z. Synthesis of the enediol isofurans, endogenous oxidation products of arachidonic acid. J. Org. Chem. 2006, 71, 926–933. [Google Scholar]
- Parker, K.A.; Lim, Y. The total synthesis of (-)-SNF4435 C and (+)-SNF4435 D. J. Am. Chem. Soc. 2004, 126, 15968–15969. [Google Scholar] [CrossRef]
- Riou, M.; Barriault, L. De novo synthesis of (+)-isofregenedol. J. Org. Chem. 2008, 73, 7436–7439. [Google Scholar]
- Yuasa, H.; Makado, G.; Fukuyama, Y. Determination of the absolute configuration of vibsanin F by asymmetric synthesis via pi-allylpalladium complex. Tetrahedron Lett. 2003, 44, 6235–6239. [Google Scholar] [CrossRef]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar]
- Takahashi, S.; Kubota, A.; Nakata, T. Stereoselective total synthesis of muconin. Tetrahedron 2003, 59, 1627–1638. [Google Scholar] [CrossRef]
- Pena, P.C.A.; Roberts, S.M. The chemistry of epoxy alcohols. Curr. Org. Chem. 2003, 7, 555–571. [Google Scholar]
- Hanson, R.M. The synthetic methodology of nonracemic glycidol and related 2,3-epoxy alcohols. Chem. Rev. 1991, 91, 437–476. [Google Scholar] [CrossRef]
- Behrens, C.H.; Sharpless, K.B. New transformations of 2,3-epoxy alcohols and related derivatives. Easy routes to homochiral substances. Aldrichim. Acta 1983, 16, 67–80. [Google Scholar]
- Rodriguez, A.; Nomen, M.; Spur, B.W.; Godfroid, J.; Lee, T.H. Total synthesis of leukotrienes from butadiene. Eur. J. Org. Chem. 2000, 2991–3000. [Google Scholar]
- Larrosa, I.; Da Silva, M.I.; Gomez, P.M.; Hannen, P.; Ko, E.; Lenger, S.R.; Linke, S.R.; White, A.J.P.; Wilton, D.; Barrett, A.G.M. Highly convergent three component benzyne coupling: The total synthesis of ent-clavilactone B. J. Am. Chem. Soc. 2006, 128, 14042–14043. [Google Scholar]
- Berberich, S.M.; Cherney, R.J.; Colucci, J.; Courillon, C.; Geraci, L.S.; Kirkland, T.A.; Marx, M.A.; Schneider, M.F.; Martin, S.F. Total synthesis of (+)-ambruticin S. Tetrahedron 2003, 59, 6819–6832. [Google Scholar]
- Heck, M.; Baylon, C.; Nolan, S.P.; Mioskowski, C. Triple ring closing metathesis reaction: Synthesis of adjacent cyclic ethers. Org. Lett. 2001, 3, 1989–1991. [Google Scholar]
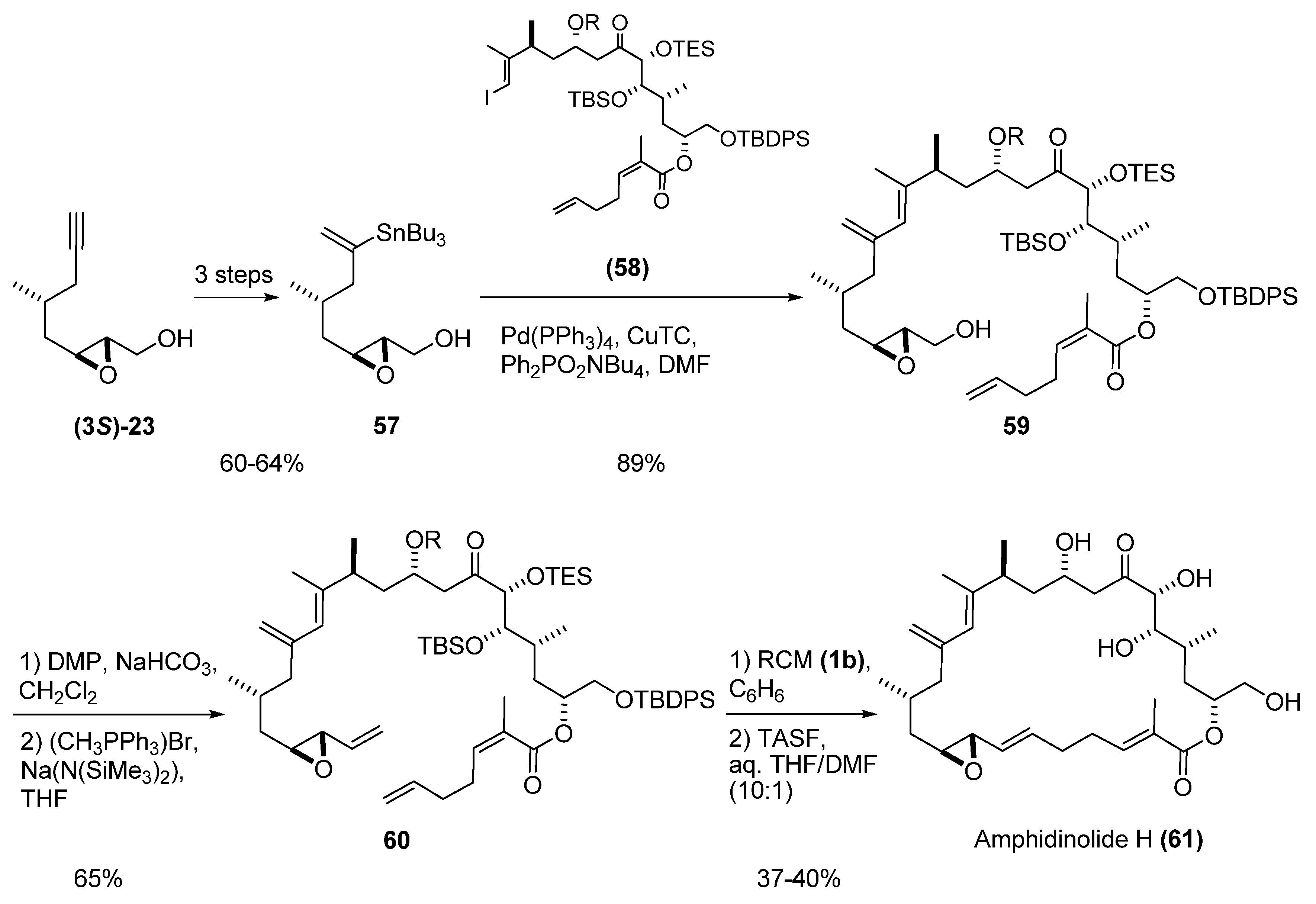
- Furstner, A.; Bouchez, L.C.; Funel, J.; Liepins, V.; Poree, F.; Gilmour, R.; Beaufils, F.; Laurich, D.; Tamiya, M. Total syntheses of amphidinolide H and G. Angew. Chem., Int. Ed. 2007, 46, 9265–9270. [Google Scholar]
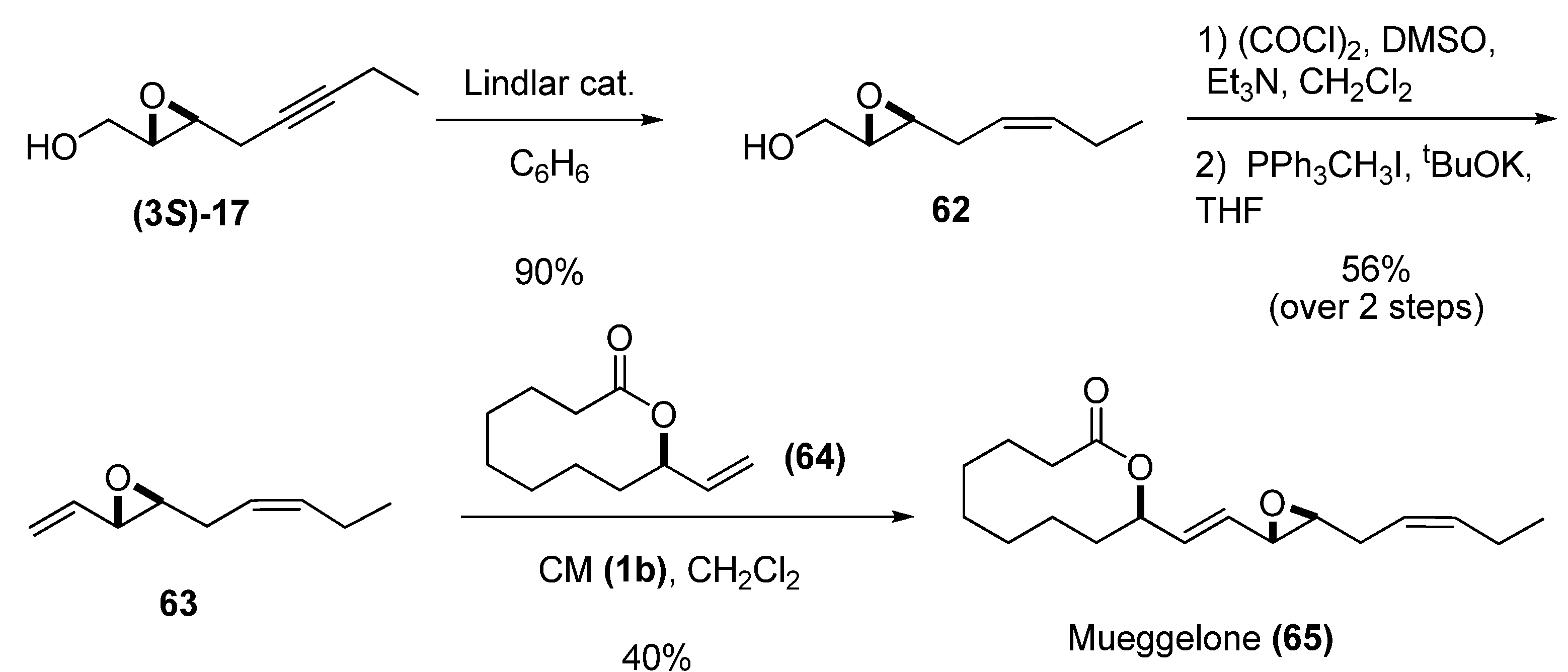
- Yadav, J.S.; Somaiah, R.; Ravindar, K.; Chandraiah, L. Stereoselective total synthesis of (+)-mueggelone, a novel inhibitor of fish development. Tetrahedron Lett. 2008, 49, 2848–2850. [Google Scholar]
- Corey, E.J.; Marfat, A.; Laguzza, B.C. Total synthesis of 5S,12S-dihydroxy-6,10-E,8,14-Z-eicosatetraenoic acid, a new human metabolite of arachidonic acid. Tetrahedron Lett. 1981, 22, 3339–3342. [Google Scholar] [CrossRef]
- Yadav, J.S.; Shekharam, T.; Gadgil, V.R. Titanocene induced regioselective deoxygenation of 2,3-epoxy alcohols: A new reaction for the synthesis of allylic alcohols. J. Chem. Soc. Chem. Commun. 1990, 843–844. [Google Scholar]
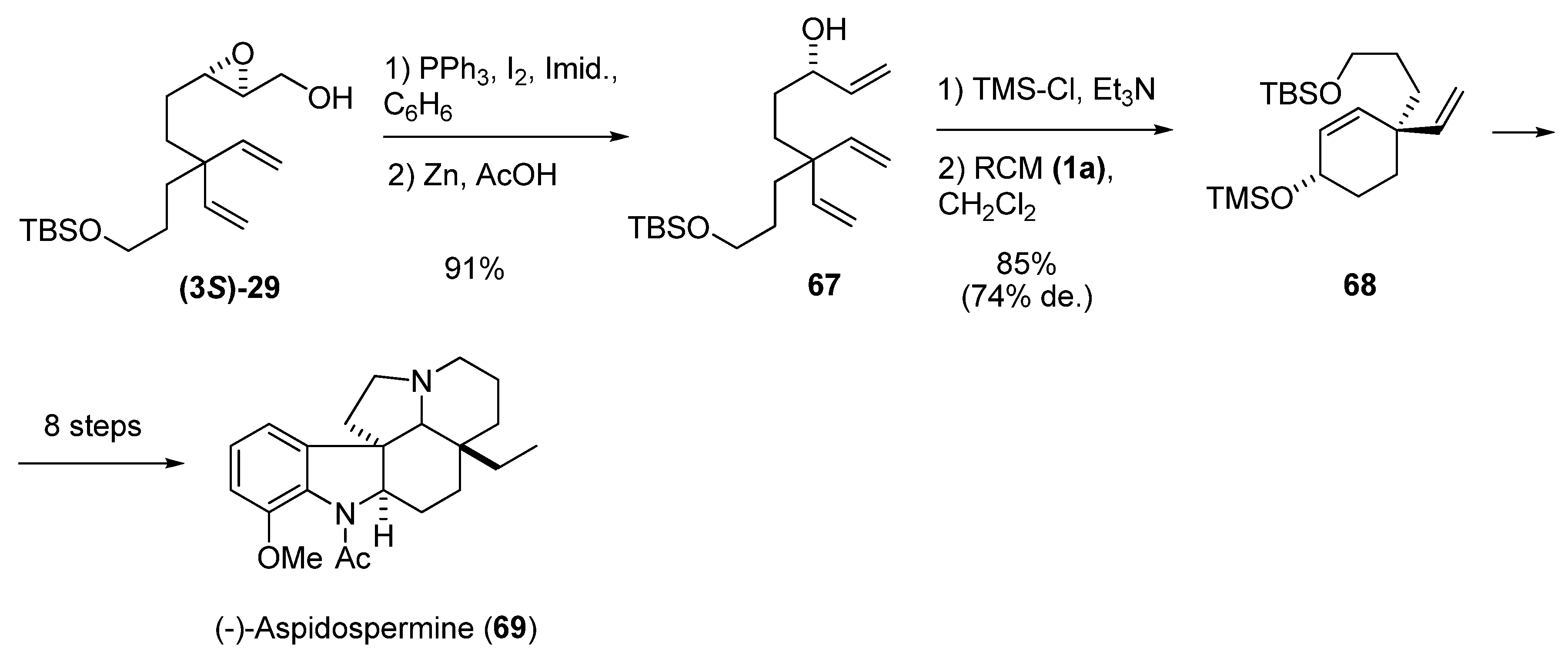
- Fukuda, Y.; Shindo, M.; Shishido, K. Total synthesis of (-)-aspidospermine via Diastereoselective ring-closing olefin metathesis. Org. Lett. 2003, 5, 749–751. [Google Scholar] [CrossRef]
- Kanada, R.M.; Itoh, D.; Nagai, M.; Niijima, J.; Asai, N.; Mizui, Y.; Abe, S.; Kotake, Y. Total synthesis of the potent antitumor macrolides pladienolide B and D. Angew. Chem. Int. Ed. 2007, 46, 4350–4355. [Google Scholar]
- Wang, Z.; Tu, Y.; Frohn, M.; Zhang, J.; Shi, Y. An efficient catalytic asymmetric epoxidation method. J. Am. Chem. Soc. 1997, 119, 11224–11235. [Google Scholar] [CrossRef]
- Sarandeses, L.A.; Mourino, A.; Luche, J.L. Cleavage of 2,3-epoxyalkyl halides by the sonochemical zinc-copper couple. J. Chem. Soc. Chem. Commun. 1991, 818–820. [Google Scholar]
- Ichige, T.; Okano, Y.; Kanoh, N.; Nakata, M. Total synthesis of methyl sarcophytoate, a marine natural biscembranoid. J. Org. Chem. 2009, 74, 230–243. [Google Scholar] [CrossRef]
- Gao, Y.; Klunder, J.M.; Hanson, R.M.; Masamune, H.; Ko, S.Y.; Sharpless, K.B. Catalytic asymmetric epoxidation and kinetic resolution: Modified procedures including in situ derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar]
- Van Dyke, A.R.; Jamison, T.F. Functionalized templates for the convergent assembly of polyethers: Synthesis of the HIJK rings of gymnocin A. Angew. Chem. Int. Ed. 2009, 48, 4430–4432. [Google Scholar] [CrossRef] [Green Version]
- Yadav, J.S.; Deshpande, P.K.; Sharma, G.V.M. An effective, practical method for the synthesis of chiral propargyl alcohols. Tetrahedron 1990, 46, 7033–7046. [Google Scholar]
- Yadav, J.S.; Bhanu, L.R.M.; Dutta, D. Stereoselective total syntheses of (9S)- and (9R)-HETE. Tetrahedron 1998, 54, 3929–3934. [Google Scholar] [CrossRef]
- Marshall, J.A.; Piettre, A.; Paige, M.A.; Valeriote, F. A modular synthesis of annonaceous acetogenins. J. Org. Chem. 2003, 68, 1771–1779. [Google Scholar]
- Hoye, T.R.; Suhadolnik, J.C. Stereocontrolled synthesis of 2,5-linked bistetrahydrofurans via the triepoxide cascade reaction. Tetrahedron 1986, 42, 2855–2862. [Google Scholar] [CrossRef]
- Kong, L.; Zhuang, Z.; Chen, Q.; Deng, H.; Tang, Z.; Jia, X.; Li, Y.; Zhai, H. A facile asymmetric synthesis of (+)-eldanolide. Tetrahedron Asymmetry 2007, 18, 451–454. [Google Scholar] [CrossRef]
- Krishna, P.R.; Lopinti, K. A concise stereoselective total synthesis of (2R,2'R)-threo-(+)-methylphenidate via a ring-closing metathesis protocol. Synlett 2007, 1742–1744. [Google Scholar]
- Ma, S.; Ni, B. Double ring-closing metathesis reaction of nitrogen-containing tetraenes: Efficient construction of bicyclic alkaloid skeletons and synthetic application to four stereoisomers of lupinine and their derivatives. Chem. Eur. J. 2004, 10, 3286–3300. [Google Scholar] [CrossRef]
- Sabitha, G.; Sudhakar, K.; Reddy, N.M.; Rajkumar, M.; Yadav, J.S. Chelation-controlled reduction: An enantioselective synthesis of (-)-tarchonanthuslactone. Tetrahedron Lett. 2005, 46, 6567–6570. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Lei, H. Chelation-controlled reduction: Stereoselective formation of syn-1,3-diols and synthesis of compactin and mevinolin lactone. J. Org. Chem. 2002, 67, 8783–8788. [Google Scholar] [CrossRef]
- Ball, M.; Bradshaw, B.J.; Dumeunier, R.; Gregson, T.J.; MacCormick, S.; Omori, H.; Thomas, E.J. A preliminary evaluation of a metathesis approach to bryostatins. Tetrahedron Lett. 2006, 47, 2223–2227. [Google Scholar]
- Hiebel, M.; Pelotier, B.; Lhoste, P.; Piva, O. Synthesis of bistramide A and analogues, part 1: Stereoselective access to normethyl tetrahydropyran subunit. Synlett 2008, 1202–1204. [Google Scholar]
- Dupradeau, F.; Prandi, J.; Beau, J. Synthesis of 2,6-dideoxy-4-S-methyl-4-thio-D-ribo-hexopyranose, a component of the esperamycin oligosaccharide. Tetrahedron 1995, 51, 3205–3220. [Google Scholar]
- Gopalarathnam, A.; Nelson, S.G. Amphidinolide, B: Asymmetric synthesis of a C7-C20 synthon. Org. Lett. 2006, 8, 7–10. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Lei, H. An enantioselective synthesis of the core unit of the non-nucleoside reverse transcriptase inhibitor taurospongin A. Tetrahedron Asymmetry 2003, 14, 629–634. [Google Scholar] [CrossRef]
- Lebel, H.; Jacobsen, E.N. Enantioselective total synthesis of taurospongin A. J. Org. Chem. 1998, 63, 9624–9625. [Google Scholar]
- Roush, W.R.; Brown, R.J. Total synthesis of carbohydrates. 3. Efficient enantioselective syntheses of 2,6-dideoxyhexoses. J. Org. Chem. 1983, 48, 5093–5101. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A. Directed openings of 2,3-epoxy alcohols via reactions with isocyanates: synthesis of (+)-erythro-dihydrosphingosine. J. Org. Chem. 1985, 50, 3752–3757. [Google Scholar]
- Martin, R.; Moyano, A.; Pericas, M.A.; Riera, A. A Concise enantioselective entry to the synthesis of deoxy-aza-sugars. Org. Lett. 2000, 2, 93–95. [Google Scholar] [CrossRef]
- Martin, R.; Murruzzu, C.; Pericas, M.A.; Riera, A. General approach to glycosidase inhibitors. Enantioselective synthesis of deoxymannojirimycin and swainsonine. J. Org. Chem. 2005, 70, 2325–2328. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Hermann, C.Y.W.; Dong, Q.; Shimizu, T.; Swaminathan, S.; Xi, N. Nitrogen heterocycles from furans. The aza-Achmatowicz reaction. Synlett 1998, 105–114. [Google Scholar]
- Shirai, M.; Okamoto, S.; Sato, F. Practical synthesis of optically active bicyclic oxazolidinylpiperidines, chiral building blocks for preparing 1-deoxyazasugars, from serine. Tetrahedron Lett. 1999, 40, 5331–5332. [Google Scholar] [CrossRef]
- Asano, K.; Hakogi, T.; Iwama, S.; Katsumura, S. New entry for asymmetric deoxyazasugar synthesis: Syntheses of deoxymannojirimycin, deoxyaltrojirimycin and deoxygalactostatin. Chem. Commun. 1999, 41–42. [Google Scholar]
- Al-Rawi, S.; Hinderlich, S.; Reutter, W.; Giannis, A. Sialic acids: Synthesis and biochemical properties of reversible inhibitors of UDP-N-acetylglucosamine 2-epimerase. Angew. Chem. Int. Ed. 2004, 43, 4366–4370. [Google Scholar] [CrossRef]
- Murruzzu, C.; Alonso, M.; Canales, A.; Jimenez-Barbero, J.; Riera, A. Synthesis and NMR experiments of (4,5,6-13C)-deoxymannojirimycin. A new entry to 13C-labeled glycosidase inhibitors. Carbohydr. Res. 2007, 342, 1805–1812. [Google Scholar] [CrossRef]
- Alegret, C.; Ginesta, X.; Riera, A. Asymmetric synthesis of cis-4- and trans-3-hydroxypipecolic acids. Eur. J. Org. Chem. 2008, 1789–1796. [Google Scholar]
- Ginesta, X.; Pericas, M.A.; Riera, A. Enantioselective synthesis of erythro-β-hydroxyglutamic acid. Synth. Commun. 2005, 35, 289–297. [Google Scholar] [CrossRef]
- Suzuki, T.; Saimoto, H.; Tomioka, H.; Oshima, K.; Nozaki, H. Regio- and stereoselective ring opening of epoxy alcohols with organoaluminum compounds leading to 1,2-diols. Tetrahedron Lett. 1982, 23, 3597–3600. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A.; Peseckis, S.M. Regioselectivity of the reactions of trialkylaluminum reagents with 2,3-epoxy alcohols: Application to the synthesis of alpha -chiral aldehydes. Tetrahedron Lett. 1983, 24, 1377–1380. [Google Scholar]
- Banwell, M.G.; McLeod, M.D.; Premraj, R.; Simpson, G.W. Improved synthetic route to enantiomerically pure samples of the tetrahydropyran-2-ylacetic acid core associated with the phytotoxic polyketide herboxidiene. Aust. J. Chem. 2000, 53, 659–664. [Google Scholar] [CrossRef]
- Balasubramaniam, R.P.; Moss, D.K.; Wyatt, J.K.; Spence, J.D.; Gee, A.; Nantz, M.H. Methylation-ring opening of 3,3-disubstituted 2,3-epoxy alcohols. Synthesis of chiral quaternary fragments for assembly of briaran diterpenes. Tetrahedron 1997, 53, 7429–7444. [Google Scholar]
- Caron, M.; Sharpless, K.B. Titanium isopropoxide-mediated nucleophilic openings of 2,3-epoxy alcohols. A mild procedure for regioselective ring-opening. J. Org. Chem. 1985, 50, 1557–1560. [Google Scholar] [CrossRef]
- Infante, I.; Bonini, C.; Lelj, F.; Righi, G. A first theoretical study on the origin of the metal-mediated regioselective opening of 2,3-epoxy alcohols. J. Org. Chem. 2003, 68, 3773–3780. [Google Scholar]
- Chini, M.; Crotti, P.; Macchia, F. Regioalternating selectivity in the metal salt catalyzed aminolysis of styrene oxide. J. Org. Chem. 1991, 56, 5939–5942. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Okazaki, M.; Miyamoto, K.; Nakata, T. Efficient phthalate-tethered ring-closing metathesis as a cross-coupling reaction. Tetrahedron Lett. 2001, 42, 7633–7636. [Google Scholar]
- McDonald, F.E.; Gleason, M.M. Asymmetric synthesis of nucleosides via molybdenum-catalyzed alkynol cycloisomerization coupled with stereoselective glycosylations of deoxyfuranose glycals and 3-amidofuranose glycals. J. Am. Chem. Soc. 1996, 118, 6648–6659. [Google Scholar] [CrossRef]
- McDonald, F.E.; Zhu, H.Y.H. Synthesis of pyranose glycals via tungsten and molybdenum pentacarbonyl-induced alkynol cyclizations. Tetrahedron 1997, 53, 11061–11068. [Google Scholar] [CrossRef]
- Poch, M.; Alcon, M.; Moyano, A.; Pericas, M.A.; Riera, A. A short enantioselective synthesis of N-Boc-α-amino acids from epoxy alcohols. Tetrahedron Lett. 1993, 34, 7781–7784. [Google Scholar] [CrossRef]
- Pasto, M.; Moyano, A.; Pericas, M.A.; Riera, A. An enantioselective, stereodivergent approach to anti- and syn-α-hydroxy-β-amino acids from anti-3-amino-1,2-diols. Synthesis of the ready for coupling Taxotere side chain. Tetrahedron Asymmetry 1996, 7, 243–262. [Google Scholar] [CrossRef]
- Pasto, M.; Castejon, P.; Moyano, A.; Pericas, M.A.; Riera, A. A catalytic asymmetric synthesis of cyclohexylnorstatine. J. Org. Chem. 1996, 61, 6033–6037. [Google Scholar]
- Pasto, M.; Moyano, A.; Pericas, M.A.; Riera, A. A catalytic asymmetric synthesis of N-Boc-β-methylphenylalanines. J. Org. Chem. 1997, 62, 8425–8431. [Google Scholar]
- Medina, E.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective syntheses of conformationally rigid, highly lipophilic mesityl-substituted amino acids. Helv. Chim. Acta 2000, 83, 972–988. [Google Scholar] [CrossRef]
- Martin, R.; Islas, G.; Moyano, A.; Pericas, M.A.; Riera, A. A new method for the enantioselective synthesis of N-Boc-α,α-disubstituted a-amino acids. Tetrahedron 2001, 57, 6367–6374. [Google Scholar] [CrossRef]
- Alcon, M.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective synthesis of unsaturated amino acids using p-methoxybenzylamine as an ammonia equivalent. Tetrahedron Asymmetry 1999, 10, 4639–4651. [Google Scholar]
- Alcon, M.; Poch, M.; Moyano, A.; Pericas, M.A.; Riera, A. Enantioselective synthesis of (S)-vigabatrin. Tetrahedron Asymmetry 1997, 8, 2967–2974. [Google Scholar]
- Martin, R.; Alcon, M.; Pericas, M.A.; Riera, A. Ring-closing metathesis of chiral allylamines. Enantioselective synthesis of (2S,3R,4S)-3,4-dihydroxyproline. J. Org. Chem. 2002, 67, 6896–6901. [Google Scholar] [CrossRef]
- Murruzzu, C.; Riera, A. Enantioselective synthesis of hydroxylated pyrrolidines via sharpless epoxidation and olefin metathesis. Tetrahedron Asymmetry 2007, 18, 149–154. [Google Scholar]
- Ginesta, X.; Pericas, M.A.; Riera, A. Straightforward entry to the pipecolic acid nucleus. Enantioselective synthesis of baikiain. Tetrahedron Lett. 2002, 43, 779–782. [Google Scholar] [CrossRef]
- Alegret, C.; Riera, A. Enantioselective synthesis of indolizidine alkaloid trans-209D. J. Org. Chem. 2008, 73, 8661–8664. [Google Scholar]
- Alegret, C.; Santacana, F.; Riera, A. Enantioselective synthesis of trans-4-methylpipecolic acid. J. Org. Chem. 2007, 72, 7688–7692. [Google Scholar] [CrossRef]
- Ginesta, X.; Pasto, M.; Pericas, M.A.; Riera, A. New stereodivergent approach to 3-amino-2,3,6-trideoxysugars. Enantioselective synthesis of daunosamine, ristosamine, acosamine, and epi-daunosamine. Org. Lett. 2003, 5, 3001–3004. [Google Scholar] [CrossRef]
- Alegret, C.; Benet-Buchholz, J.; Riera, A. Stereodivergent syntheses of conduramines and aminocyclitols. Org. Lett. 2006, 8, 3069–3072. [Google Scholar] [CrossRef]
- Sample Availability: Contact the authors.
© 2010 by the authors;
Share and Cite
Riera, A.; Moreno, M. Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation. Molecules 2010, 15, 1041-1073. https://doi.org/10.3390/molecules15021041
Riera A, Moreno M. Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation. Molecules. 2010; 15(2):1041-1073. https://doi.org/10.3390/molecules15021041
Chicago/Turabian StyleRiera, Antoni, and María Moreno. 2010. "Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation" Molecules 15, no. 2: 1041-1073. https://doi.org/10.3390/molecules15021041
APA StyleRiera, A., & Moreno, M. (2010). Synthetic Applications of Chiral Unsaturated Epoxy Alcohols Prepared by Sharpless Asymmetric Epoxidation. Molecules, 15(2), 1041-1073. https://doi.org/10.3390/molecules15021041