Synthesis of syn-γ-Amino-β-hydroxyphosphonates by Reduction of β-Ketophosphonates Derived from L-Proline and L-Serine

Abstract
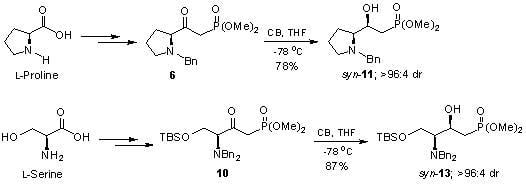
:
1. Introduction

2. Results and Discussion



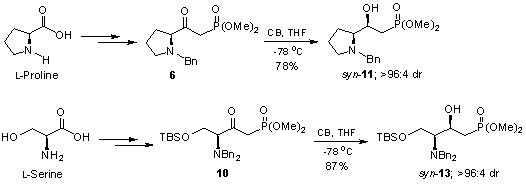{kind=link}
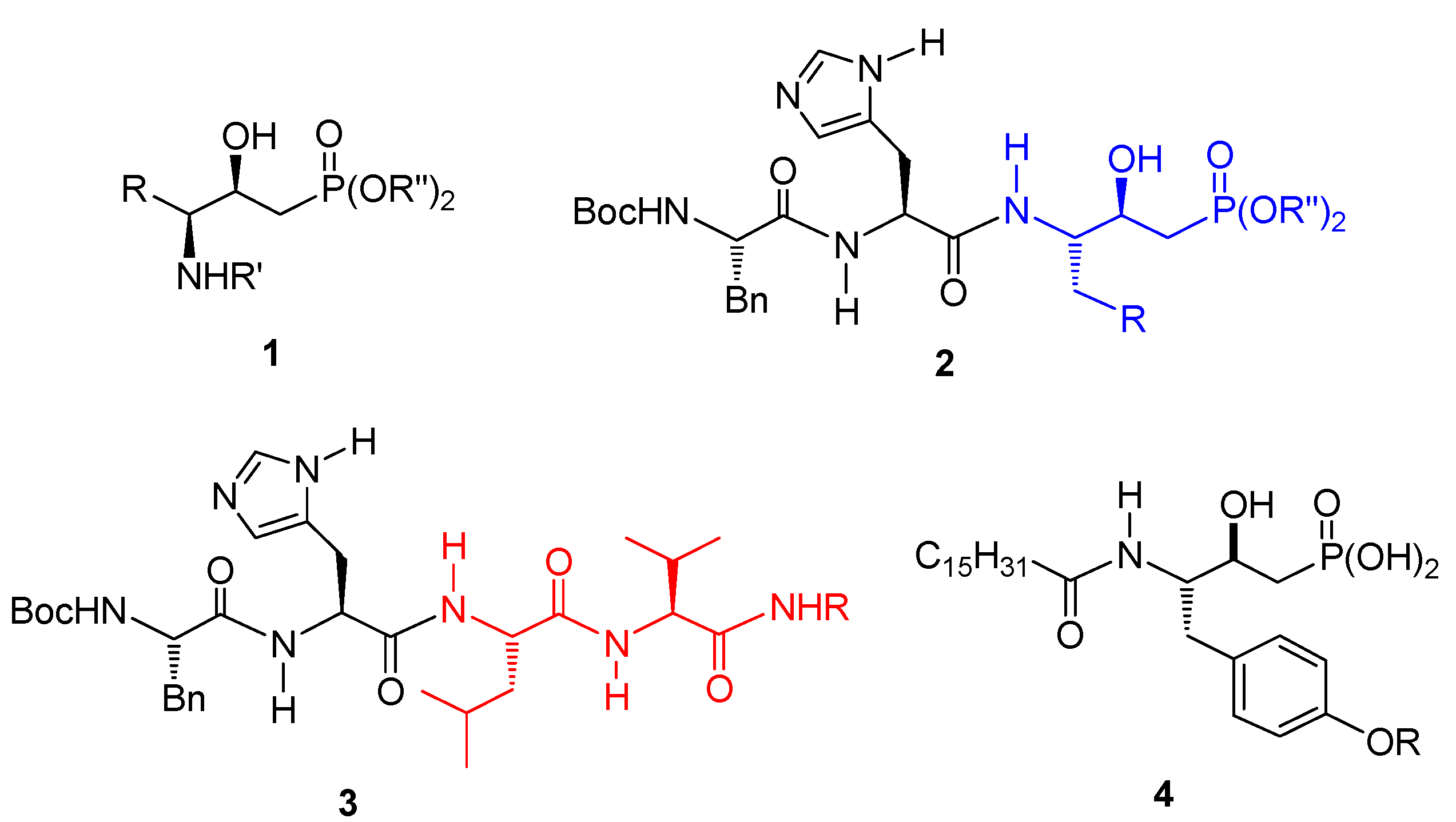{kind=link}
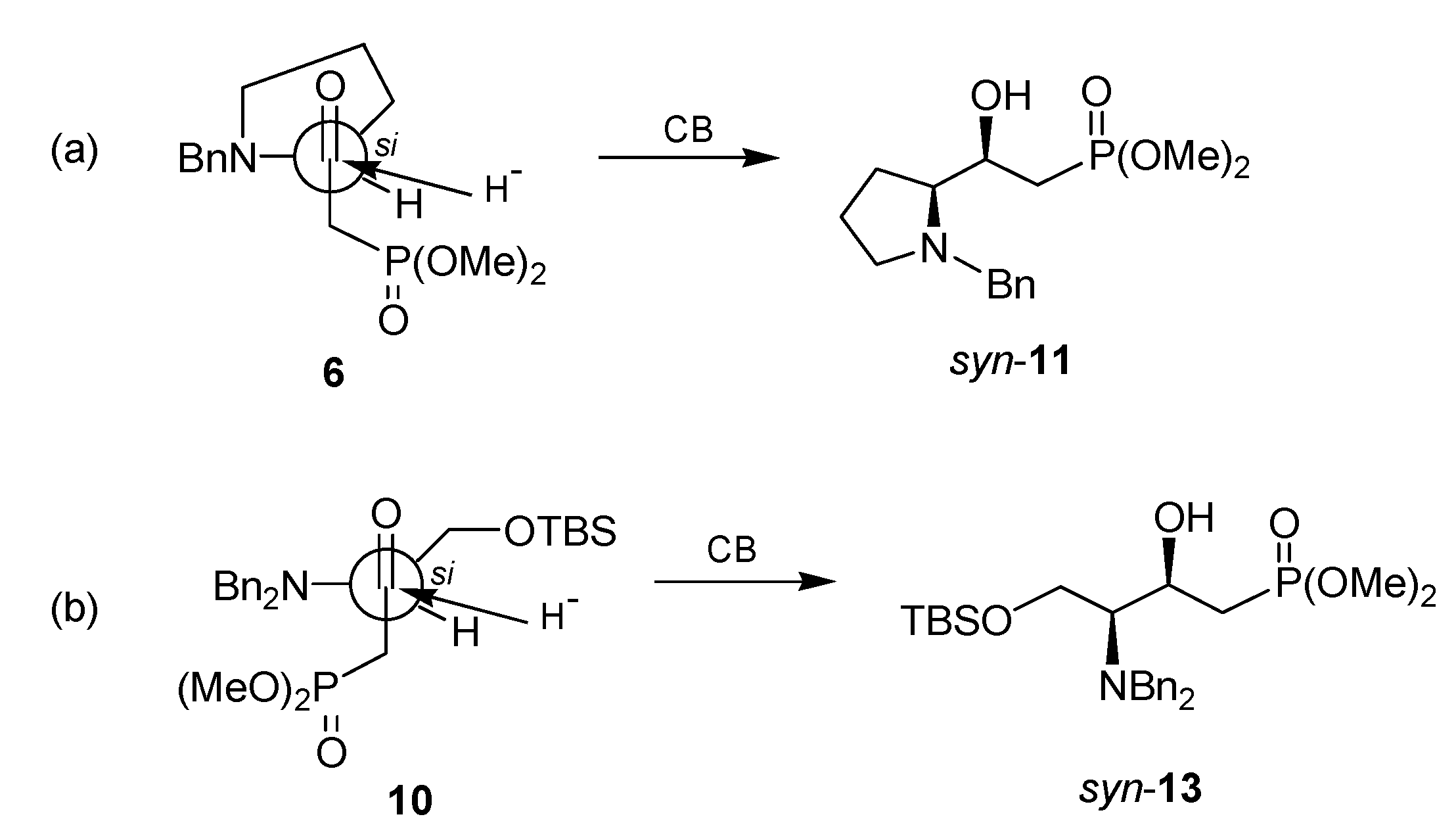{kind=link}
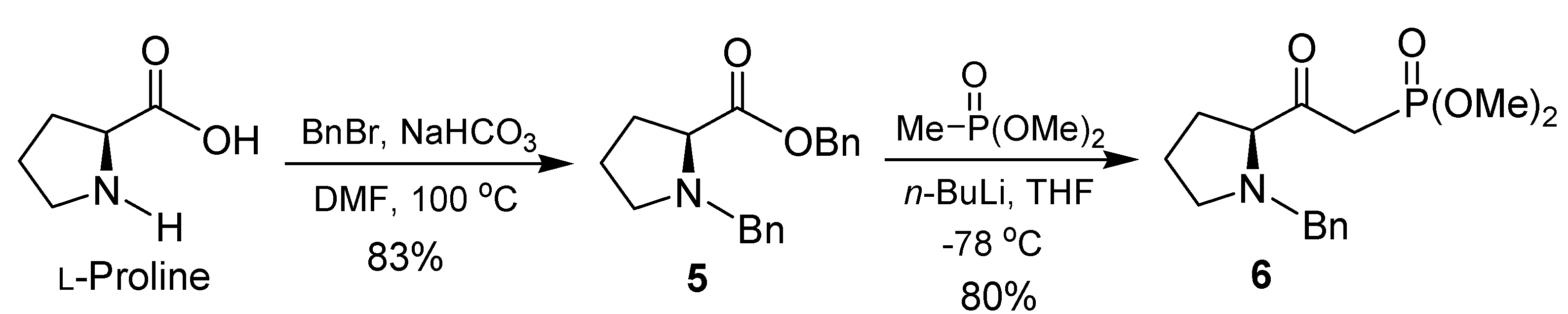{kind=link}
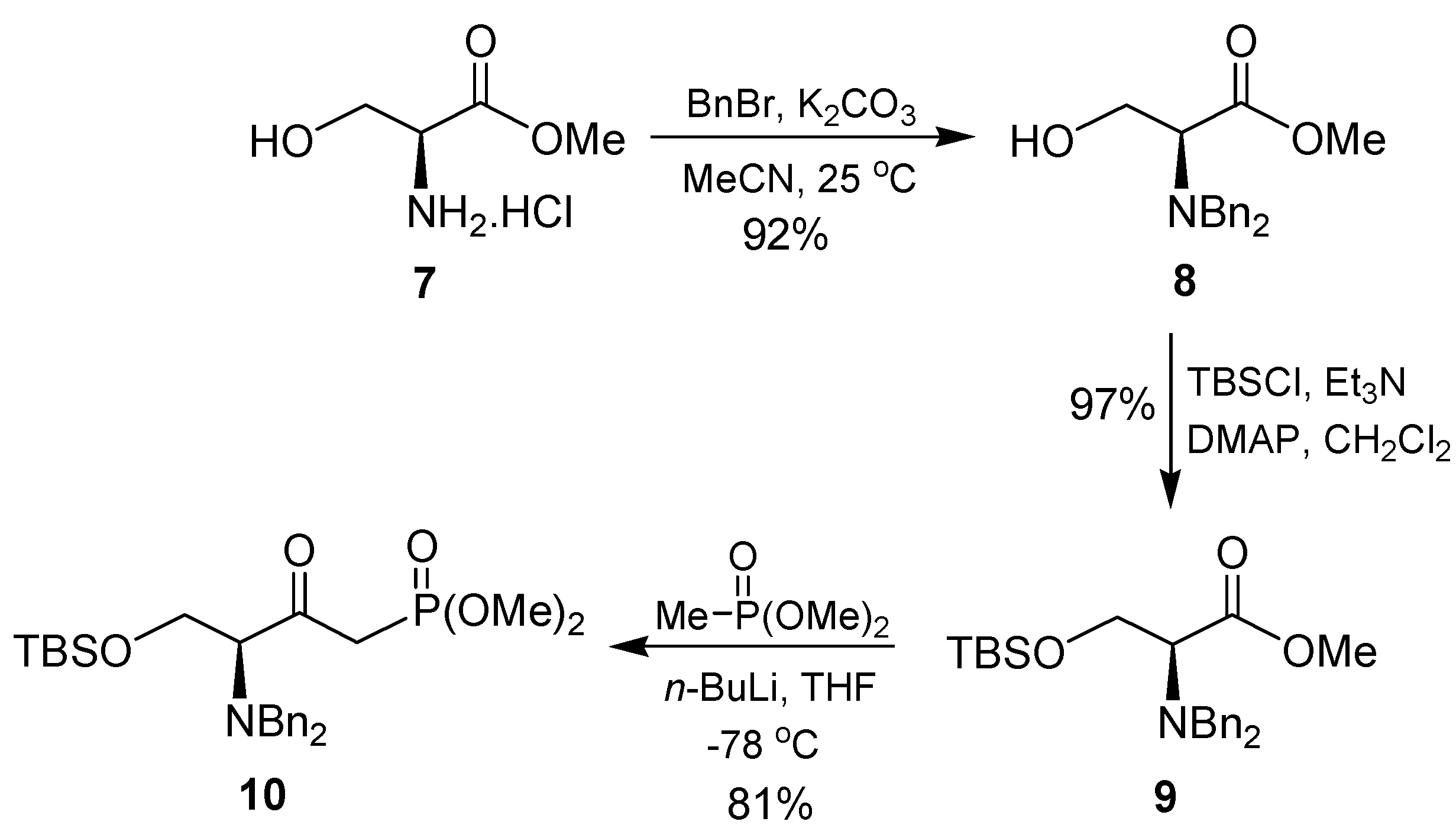{kind=link}
 | |||||
| Entry | Hydride | Conditions | Yield (%)a | syn- 11: anti- 12 b | |
| 1 | NaBH4 | MeOH, 25 °C | 70 | 69:31 | |
| 2 | LiBH4 | THF, -78 °C | 69 | 75:25 | |
| 3 | DIBAL-H | THF, -78 °C | 69 | 79:21 | |
| 4 | CB | THF, -78 °C | 78 | ≥96:4 | |
 | ||||
| Entry | Hydride | Conditions | Yield (%)a | syn-13: anti-14b |
| 1 | NaBH4 | MeOH, 25 °C | 70 | 81:19 |
| 2 | LiBH4 | THF, -78 °C | 75 | 82:18 |
| 3 | DIBAL-H | THF, -78 °C | 91 | 88:12 |
| 4 | CB | THF, -78 °C | 87 | ≥96:4 |

3. Experimental
3.1. General Procedures
4. Conclusions
Acknowledgements
- Samples Availability: Samples of the compounds are available from authors.
References and Notes
- Ordóñez, M.; Rojas-Cabrera, H.; Cativiela, C. An Overview of Stereoselective Synthesis of α-Aminophosphonic Acids and Derivatives. Tetrahedron 2009, 65, 17–49, For a review on the stereoselective synthesis of α-aminophosphonic acids and derivatives, see. [Google Scholar]
- Aminophosphonic and Aminophosphinic Acids; Kukhar, V.P.; Hudson, H.R. (Eds.) John Wiley & Sons: Chichester, UK, 2000.
- Kafarsky, P.; Lejczak, B. Biological Activity of Aminophosphonic Acids. Phosphorous, Sulfur Silicon 1991, 63, 193–215. [Google Scholar]
- McLeod, D.A.; Brinkworth, R.I.; Ashley, J.A.; Janda, K.D.; Wirsching, P. Phosphonamidates and Phosphonamidate Esters as HIV-1 Protease Inhibitors. Bioorg. Med. Chem. Lett. 1991, 1, 653–658. [Google Scholar] [CrossRef]
- Nieschalk, J.; Batsanov, A.S.; O’Hagan, D.; Howard, J.A.K. Synthesis of Monofluoro- and Difluoromethylenephosphonate Analogues of sn-glycerol-3-phosphate as Substrate for Glycerol-3-phosphate Dehydrogenase and the X-ray Structure of the Fluoromethylene-phosphonate Moiety. Tetrahedron 1996, 52, 165–176. [Google Scholar]
- Burke, T.R., Jr.; Smyth, M.S.; Nomizu, M.; Otaka, A. Roller, P.P. Preparation of Fluoro- and Hydroxy-4-(phosphonomethyl)-D,L-phenylalanine Suitable Protected for Solid-Phase Synthesis of Peptides containing Hydrolytically Stable Analogs of O-phosphotyrosine. J. Org. Chem. 1993, 58, 1336–1340. [Google Scholar]
- Dellaria, J.F., Jr.; Maki, R.G.; Stein, H.H.; Cohen, J.; Whittern, D.; Marsh, K.; Hoffman, D.J.; Plattner, J.J.; Perun, T.J. New Inhibitors of Renin that Contain Novel Phosphostatine Leu-Val replacements. J. Med. Chem. 1990, 33, 534–542. [Google Scholar] [CrossRef]
- Chakravarty, P.K.; Greenlee, W.J.; Parsons, W.H.; Patchett, A.A.; Combs, P.; Roth, A.; Busch, R.D.; Mellin, T.N. (3-Amino-2-oxoalkyl)phosphonic Acids and Their Analogs as Novel Inhibitors of D-Alanine:D-alaline Ligase. J. Med. Chem. 1989, 32, 1886–1890. [Google Scholar] [CrossRef]
- Cui, P.; McCalmont, W.F.; Tomsig, J.L.; Lynch, K.R.; MacDonald, T.L. α- and β-Substituted phosphonate analogs of LPA as Autotoxin Inhibitors. Bioorg. Med. Chem. 2008, 16, 2212–2225. [Google Scholar] [CrossRef]
- Hale, J.J.; Neway, W.; Mills, S.G.; Hajdu, R.; Keohane, C.A.; Rosenbach, M.; Milligan, J.; Shein, G.J.; Chrebet, G.; Bergstrom, J.; Card, D.; Koo, G.C.; Koprak, S.L.; Jackson, J.J.; Rosen, H.; Mandala, S. Potent S1P Receptor Agonists Replicate the Pharmacologic Actions of the Novel Immune Modulator FTY720. Bioorg. Med. Chem. Lett. 2004, 14, 3351–3355. [Google Scholar]
- Chang, R.; Vo, T.-T.; Finney, N.S. Synthesis of the C1-Phosphonate Analog of UDP-GlcNAc. Carbohydr. Res. 2006, 341, 1998–2004. [Google Scholar]
- Torres-Sánchez, M.I.; Zaccaria, C.; Buzzi, B.; Miglio, G.; Lombardi, G.; Polito, L.; Russo, G.; Lay, L. Synthesis and Biological Evaluation of Phosphono Analogues of Capsular Polysaccharide Fragments from Neisseria meningitidis A. Chem. A. Eur. J. 2007, 13, 6623–6635. [Google Scholar]
- Kolodiazhnyi, O.I. Asymmetric Synthesis of Hydroxyphosphonates. Tetrahedron: Asymmetry 2005, 16, 3295–3340, and references therein. [Google Scholar]
- Bowery, N.G.; Bettler, B.; Froestl, W.; Gallagher, J.P.; Marshall, F.; Raiteri, M.; Bonner, T.I.; Enna, S.J. International Union of Pharmacology. XXXIII. Mammalian γ-Aminobutyric AcidB Receptors: Structure and Function. Pharmacol. Rev. 2002, 54, 247–264. [Google Scholar]
- Pozza, M.F.; Manuel, N.A.; Steinmann, M.; Froestl, W.; Davies, C.H. Comparison of Antagonist Potencies at Pre- and Post-synaptic GABAB receptors at Inhibitory Synapses in the CA1 Region of the Rat Hippocampus. Br. J. Pharmacol. 1999, 127, 211–219. [Google Scholar] [CrossRef]
- Tadeusiak, E.; Krawiecka, B.; Michalski, J. Synthesis of Phosphocarnitine. Tetrahedron Lett. 1999, 40, 1791–1792. [Google Scholar] [CrossRef]
- Yuan, C.-Y.; Wang, K.; Li, Z.-Y. Enantioselective Reduction of 2-Keto-3-haloalkane Phosphonates by Baker´s Yeast. Heteroat. Chem. 2001, 12, 551–556. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Łuczak, J.; Kiełbasinski, P. Chemoenzymatic Synthesis of Phosphocarnitine Enantiomers. J. Org. Chem. 2002, 67, 7872–7875. [Google Scholar]
- Wróblewski, A.E.; Hałajewska-Wosik, A. An Efficient Synthesis of Enantiomeric (S)-Phosphocarnitine. Eur. J. Org. Chem. 2002, 16, 2758–2763. [Google Scholar]
- Yuan, C.-y.; Wang, K.; Li, J.; Li, Z.-Y. Stereoselective Synthesis of Phosphorus Analogs of (R)-Carnitine and (R)-GABOB. Phosphorous, Sulfur Silicon 2002, 177, 2391–2397. [Google Scholar]
- Wang, K.; Zhang, Y.; Yuan, C. Enzymatic Synthesis of Phosphocarnitine, Phosphogabob and Fosfomycin. Org. Biomol. Chem. 2003, 1, 3564–3569. [Google Scholar] [CrossRef]
- Ordóñez, M.; González-Morales, A.; Ruíz, C.; De la Cruz-Cordero, R.; Fernández-Zertuche, M. Preaparation of (R)- and (S)-γ-amino-β-hydroxypropylphosphonic Acid from Glycine. Tetrahedron: Asymmetry 2003, 14, 1775–1779. [Google Scholar]
- Wróblewski, A.E.; Hałajewska-Wosik, A. An Efficient Synthesis of an Enantiomerically Pure Phosphonate Analogue of L-GABOB. Tetrahedron: Asymmetry 2003, 14, 3359–3363. [Google Scholar]
- Ordóñez, M.; González-Morales, A.; Salazar-Fernández, H. Highly Diastereoselective Reduction of β-Ketophosphonates Bearing Homochiral Bis-(α-methylbenzyl)amine: Preaparation of Both Enantiomers of Phosphogabob (GABOBP). Tetrahedron Asymmetry 2004, 15, 2719–2725. [Google Scholar]
- Nesterov, V.V.; Kolodyazhnyi, O.I. Enantioselective Reduction of Ketophosphonates Using Chiral Acid Adducts with Sodium Borohydride. Russ. J. Gen. Chem. 2006, 76, 1022–1030. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. New Method for the Asymmetric Hydroboration of Ketophosphonates and the Synthesis of Phosphocarnitine. Tetrahedron Asymmetry 2006, 17, 1023–1026. [Google Scholar] [CrossRef]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Efficient Method for the Asymmetric Reduction of α- and β-Ketophosphonates. Tetrahedron 2007, 63, 6720–6731. [Google Scholar]
- Nesterov, V.V.; Kolodiazhnyi, O.I. Di(1R,2S,5R)-menthyl 2-Hydroxy-3-chloropropylphosphonates as a Useful Chiral Synthon for the Preparation of Enantiomerically Pure Phosphonic Acids. Synlett 2007, 15, 2400–2404. [Google Scholar]
- Vargas, S.; Suárez, A.; Álvarez, E.; Pizzano, A. Highly Enantioselective Hydrogenation of Enol Ester Phosphonates: A Versatile Procedure for the Preparation of Chiral β-Hydroxyphosphonates. Chem. Eur. J. 2008, 14, 9856–9859. [Google Scholar]
- Ciardi, C.; Romerosa, A.; Serrano-Ruiz, M.; Gonsalvi, L.; Peruzzini, M.; Reginato, G. Synthesis of New Enantiomerically enriched β-Hydroxy-γ-amino Phosphines by Selective Transformation of Naturally Occurring Amino Acids. J. Org. Chem. 2007, 72, 7787–7789. [Google Scholar]
- Clayden, J.; Collington, E.W.; Warren, S. Stereocontrolled Synthesis of R or S, E or Z-Unsaturated α-Amino Acids by Enantio- and Diastereoselective Epoxidation of δ-Hydroxy Allylic Phosphine Oxides. Tetrahedron Lett. 1993, 34, 1327–1330. [Google Scholar]
- Clayden, J.; McElroy, A. B.; Warren, S. Control Over Absolute (R,S), Relative (syn,anti) and Geometrical (E,Z) Stereochemistry in the Synthesis of Allylcally Substituted Alkenes from Diphenylphosphinoyl Epoxy Alcohols. J. Chem. Soc., Perkin Trans. 1 1995, 13, 1913–1934. [Google Scholar]
- Krawiecka, B.; Jerziorna, A. Stereocontrolled Synthesis of 3-Amino-2-hydroxyalkyl Diphenylphosphine Oxides Mediated by Chiral Azetidinium Salts and Epoxyamines. Tetrahedron Lett. 2005, 46, 4381–4384. [Google Scholar] [CrossRef]
- Dellaria, J.F.; Maki, R.G. The Enantio- and Diastereoselective Synthesis of the First Phospho-statine Derivative. Tetrahedron Lett. 1986, 27, 2337–2340. [Google Scholar] [CrossRef]
- Shalem, H.; Shtzamiller, S.; Feit, B.-A. Synthesis of 2-(Aminophenyl)-2-hydroxyethyl-phosphonates and Their Incorporation in Short Peptides. Liebigs Ann. 1995, 433–436. [Google Scholar]
- Froestl, W.; Mickel, S.J.; Hall, R.G.; Sprecher, G.V.; Strub, D.; Baumman, P.A.; Brugger, F.; Gentsch, C.; Jaeckel, J.; Olpe, H.-R.; Rihs, G.; Vassout, A.; Waldmeier, P.C.; Bittiger, H. Phosphinic Acid Analogs of GABA. 1. New Potent and Selective GABAB Agonists. J. Med. Chem. 1995, 38, 3297–3312. [Google Scholar]
- Otaka, A.; Miyoshi, K.; Burke, T.R., Jr.; Roller, P.P.; Kubota, H.; Tamamura, H.; Fujii, N. Synthesis and Application of N-Boc-L-Amino-4-(diethylphosphono)-4,4-difluorobutanoic Acid for Solid-Phase Synthesis of Nonhydrolyzable Phosphoserine Peptide Analogues. Tetrahedron Lett. 1995, 36, 927–930. [Google Scholar]
- Yokomatsu, T.; Takechi, H.; Akiyama, T.; Shibuya, S.; Kominato, T.; Soeda, S.; Shimeno, H. Synthesis and Evaluation of Difluoromethylene Analogue of Sphingomyelin as an Inhibitor of Sphingomyelinase. Bioorg. Med. Chem. Lett. 2001, 11, 1277–1280. [Google Scholar]
- Yokomatsu, T.; Murano, T.; Akiyama, T.; Koizumi, J.; Shibuya, S.; Tsuji, Y.; Soeda, S.; Shimeno, H. Synthesis of Non-competitive Inhibitors of Sphingomyelinases with Significant Activity. Bioorg. Med. Chem. Lett. 2003, 13, 229–236. [Google Scholar]
- O’Brien, P.; Warren, S. Synthesis of Phenylalanine-derivative β-Hydroxy and β-Keto Phosphine Oxides - Investigation of the Configurational Stability of Lithiated Phosphine Oxides Using the Hoffmann Test. Tetrahedron Lett. 1996, 37, 4271–4274. [Google Scholar] [CrossRef]
- O’Brien, P.; Powell, H.R.; Raithby, P.R.; Warren, S. Investigation of the Configurational Stability of Lithiated Phosphine Oxides Using the Hoffmann Tetst: X-ray Structures of (2S*,3S*,4R*)-2-(N,N-Dibenz-ylamino)-4-diphenylphosphinoyl-1-phenylpentan-3-ol and (2S*,4S*)-2-(N,N-Dibenzylamino-4-diphenyl-phosphinoyl-1-phenylpentan-3-one. J. Chem. Soc., Perkin Trans. 1 1997, 7, 1031–1039. [Google Scholar]
- Thomas, A.A.; Sharpless, K.B. The Catalytic Asymmetric Aminohydroxylation of Unsaturated Phosphonates. J. Org. Chem. 1999, 64, 8379–8385. [Google Scholar] [CrossRef]
- Yamagishi, T.; Fujii, K.; Shibuya, S.; Yokomatsu, T. Asymmetric Synthesis of Phosphonic Acids Analogues for Acylcarnitine. Tetrahedron 2006, 62, 54–65. [Google Scholar]
- Yamagishi, T.; Fujii, K.; Shibuya, S.; Yokomatsu, T. Synthesis of Chiral γ-Amino-β-hydroxyphosphonate Derivatives from Unsaturated Phosphonates. Synlett 2004, 14, 2505–2508. [Google Scholar]
- Chen, S.; Yuan, C. Studies on Organophosphorus Compounds 82. Synthesis of Some Functionalized Difluoromethylphosphonates. Phosphorous, Sulfur Silicon 1993, 82, 73–78. [Google Scholar]
- Berkowitz, D.B.; Eggen, M.-J.; Shen, Q.; Shoemaker, R.K. Ready Access to Fluorinated Phosphonate Mimics of Secondary Phosphates. Synthesis of the (α,α-Difluoroalkyl)phosphonate Analogues of L-Phosphoserine, L-Phosphoallotreonine, and L-Phosphothreonine. J. Org. Chem. 1996, 61. [Google Scholar]
- Cui, P.; Tomsig, J.L.; McCalmont, W.F.; Lee, S.; Becker, Ch.J.; Lynch, K.R.; MacDonald, T.L. Synthesis and Biological Evaluation of phosphonate derivative as Autotaxin (ATX) Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 1634–1640. [Google Scholar] [CrossRef]
- De la Cruz-Cordero, R.; Hernández-Núñez, E.; Fernández-Zertuche, M.; Muñoz-Hernández, M.A.; Ordóñez, M. Preparation of Phosphostatine and Phosphoepistatine from L-Leucine via High Diastereoselective Reduction of 3-Amino-2-ketophosphonates. ARKIVOK 2005vi, 277–286.
- De la Cruz-Cordero, R.; Labastida-Galván, V.; Fernández-Zertuche, M.; Ordóñez, M. Preparation of Phosphostatine Analogues from L-Amino Acids. J. Mex. Chem. Soc. 2005, 49, 312–321. [Google Scholar]
- Ordóñez, M.; De la Cruz-Cordero, R.; Fernández-Zertuche, M.; Muñoz-Hernández, M.A. Diastereoselective Reduction of γ-N,N-Dibenzylamino-β-ketophosphonates γ-Substituted. Tetrahedron: Asymmetry 2002, 13, 559–562. [Google Scholar]
- Ordóñez, M.; De la Cruz-Cordero, R.; Fernández-Zertuche, M.; Muñoz-Hernández, M.A.; García-Barradas, O. Diastereoselective Reduction of Dimethyl γ-[(N-p-Toluenesulfonyl)-amino]-β-ketophosphonates Derived from Amino Acids. Tetrahedron Asymmetry 2004, 15, 3035–3043. [Google Scholar] [CrossRef]
- Ordóñez, M.; De la Cruz-Cordero, R.; Quiñones, C.; González-Morales, A. Highly Diastereoselective Synthesis of anti-γ-N-Benzylamino-β-hydroxyphosphonates. Chem. Commun. 2004, 6, 672–673. [Google Scholar]
- Goldstein, S.W.; Overman, L.E.; Rabinowitz, M.H. The First Enantioselective Total Synthesis of the Allopumiliotoxin A Alkaloids 267A and 339B. J. Org. Chem. 1992, 57, 1179–1190. [Google Scholar] [CrossRef]
- Corey, E.J.; Margriotis, P.A. Total Synthesis and Absolute Configuration of 7,20-Diisocyanoadociane. J. Am. Chem. Soc. 1987, 109, 287–289. [Google Scholar] [CrossRef]
- Cherest, M.; Felkin, H.; Prudent, N. Torsional Strain Involving Partial Bonds. The Stereochemistry of the Lithium Aluminium Hydride Reduction of Some Simple Open-chain Ketones. Tetrahedron Lett. 1968, 18, 2199–2204. [Google Scholar]
- Cherest, M.; Felkin, H. Torsional Strain Involving Partial Bonds. The Steric Course of the Reaction Between Allyl Magnesium Bromide and 4-t-Butyl-cyclohexanone. Tetrahedron Lett. 1968, 18, 2205–2208. [Google Scholar]
- Anh, N.T.; Eisenstein, O. Theoretical Interpretation of 1,2 Asymmetric Induction – Importance of Antiperiplanarity. Nouv. J. Chem. 1977, 1, 61–70. [Google Scholar]
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; John Wiley & Sons: New York, NY, USA, 1994; p. 876, For an excellent summary, see. [Google Scholar]
- Läib, T.; Chastanet, J.; Zhu, J. Diastereoselective Synthesis of γ-Hydroxy-β-amino Alcohols and (2S,3S)-β-Hydroxyleucine from Chiral D-(N,N-Dibenzylamino)serine (TBDMS). J. Org. Chem. 1998, 63, 1709–1713. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ordóñez, M.; Lagunas-Rivera, S.; Hernández-Núñez, E.; Labastida-Galván, V. Synthesis of syn-γ-Amino-β-hydroxyphosphonates by Reduction of β-Ketophosphonates Derived from L-Proline and L-Serine. Molecules 2010, 15, 1291-1301. https://doi.org/10.3390/molecules15031291
Ordóñez M, Lagunas-Rivera S, Hernández-Núñez E, Labastida-Galván V. Synthesis of syn-γ-Amino-β-hydroxyphosphonates by Reduction of β-Ketophosphonates Derived from L-Proline and L-Serine. Molecules. 2010; 15(3):1291-1301. https://doi.org/10.3390/molecules15031291
Chicago/Turabian StyleOrdóñez, Mario, Selene Lagunas-Rivera, Emanuel Hernández-Núñez, and Victoria Labastida-Galván. 2010. "Synthesis of syn-γ-Amino-β-hydroxyphosphonates by Reduction of β-Ketophosphonates Derived from L-Proline and L-Serine" Molecules 15, no. 3: 1291-1301. https://doi.org/10.3390/molecules15031291
APA StyleOrdóñez, M., Lagunas-Rivera, S., Hernández-Núñez, E., & Labastida-Galván, V. (2010). Synthesis of syn-γ-Amino-β-hydroxyphosphonates by Reduction of β-Ketophosphonates Derived from L-Proline and L-Serine. Molecules, 15(3), 1291-1301. https://doi.org/10.3390/molecules15031291