Asymmetric Ruthenium(II) and Osmium(II) Complexes with New Bidentate Polyquinoline Ligands. Synthesis and NMR Characterization

Abstract
:1. Introduction
| Ligand | R1 | Nomenclature | Initials |
| L1 | –OCH3 | 4-p-methoxyphenyl-6-bromo-2-(2′-pyridyl)quinoline | mphbr-pq |
| L2 | –OH | 4-p-hydroxyphenyl-6-bromo-2-(2′-pyridyl)quinoline | hphbr-pq |
2. Result and Discussion
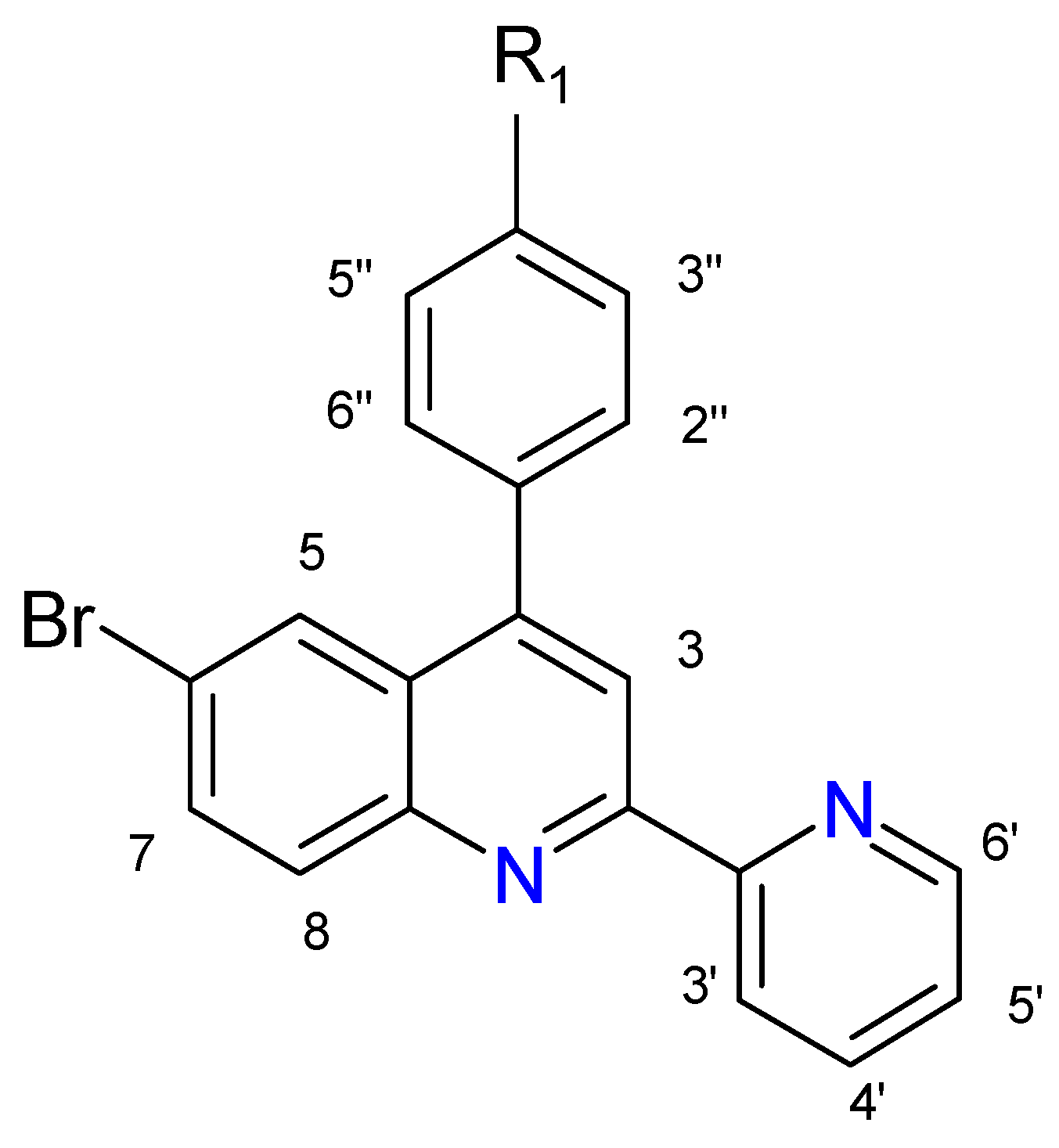
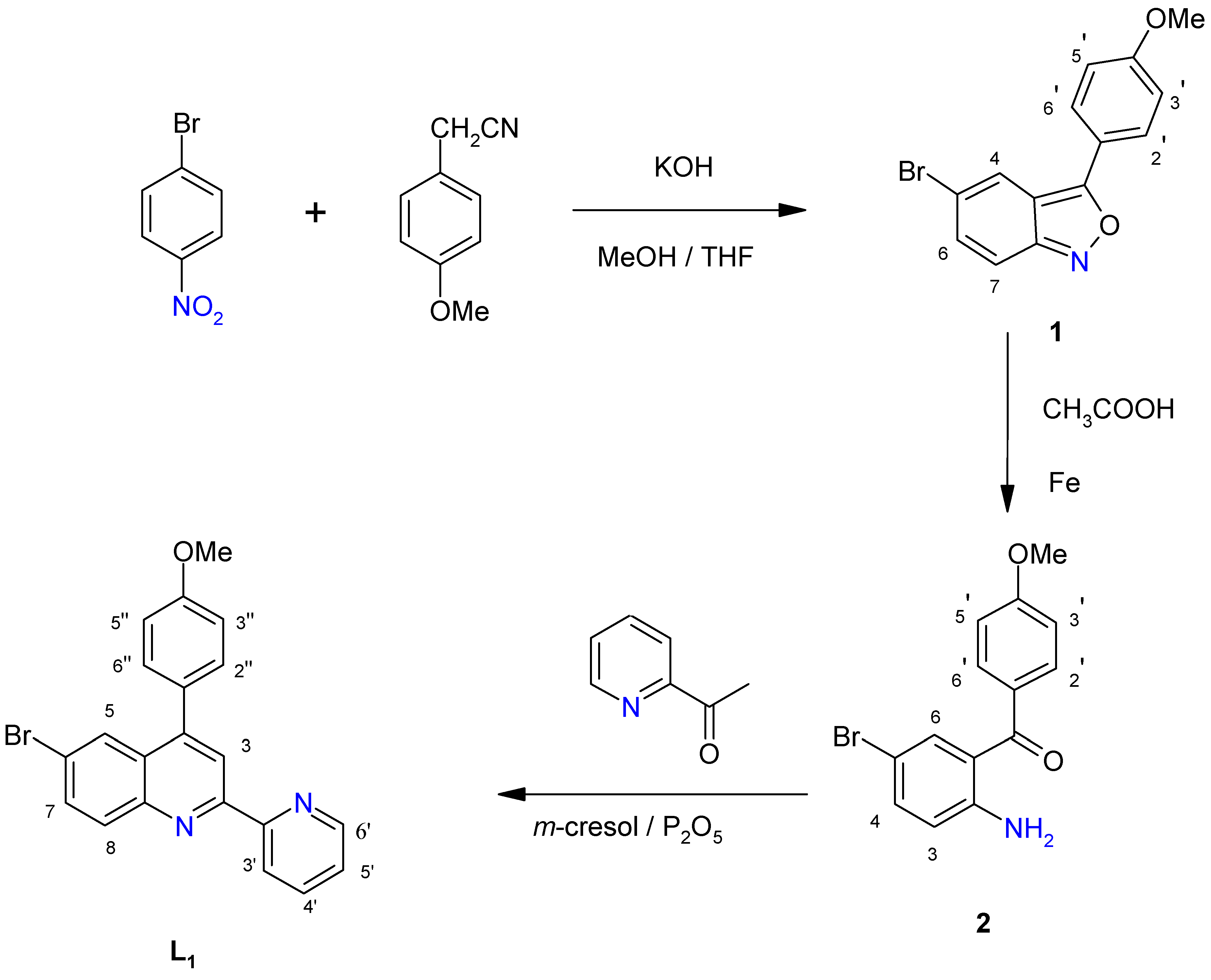
2.1. Ligands
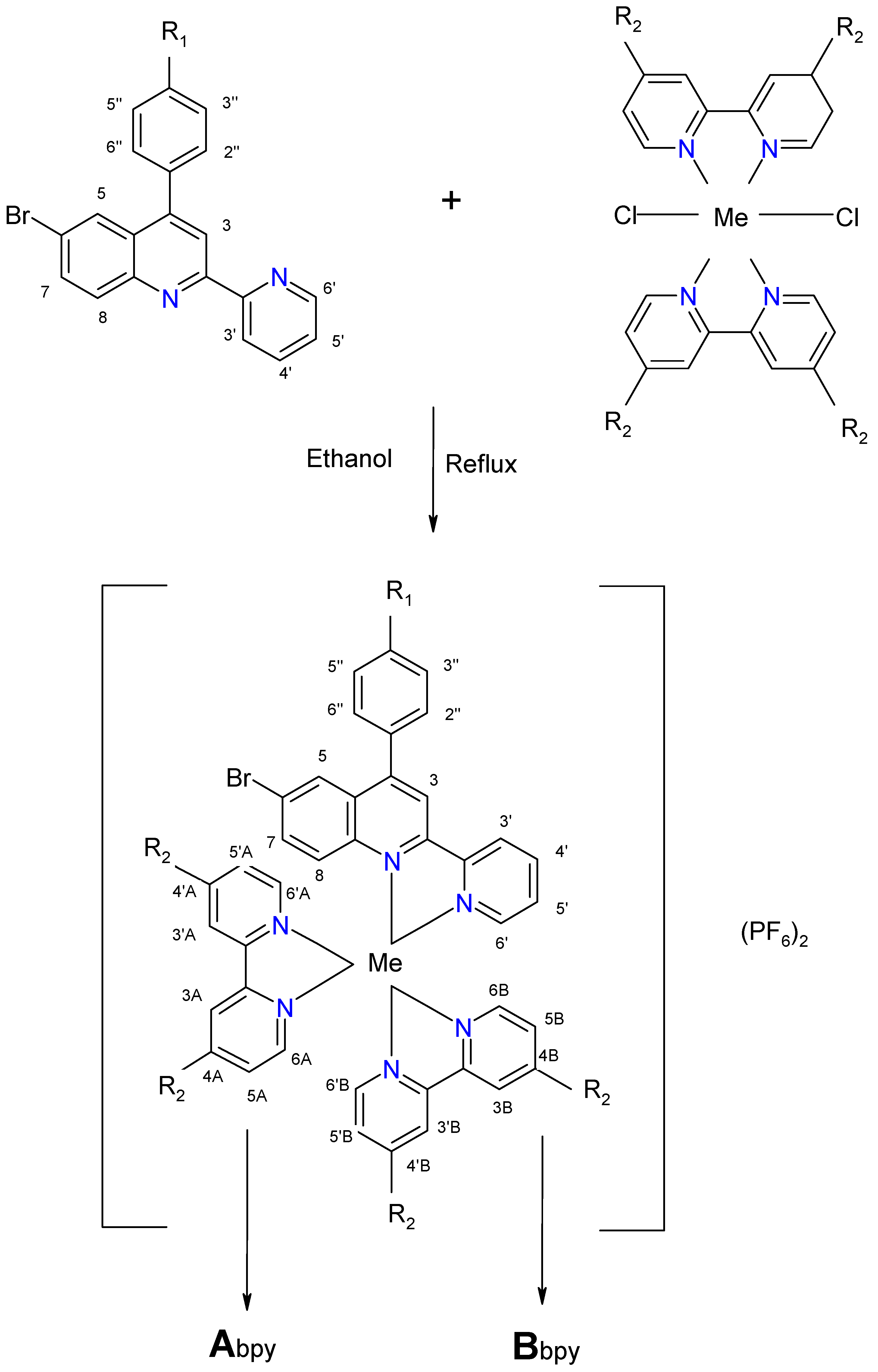
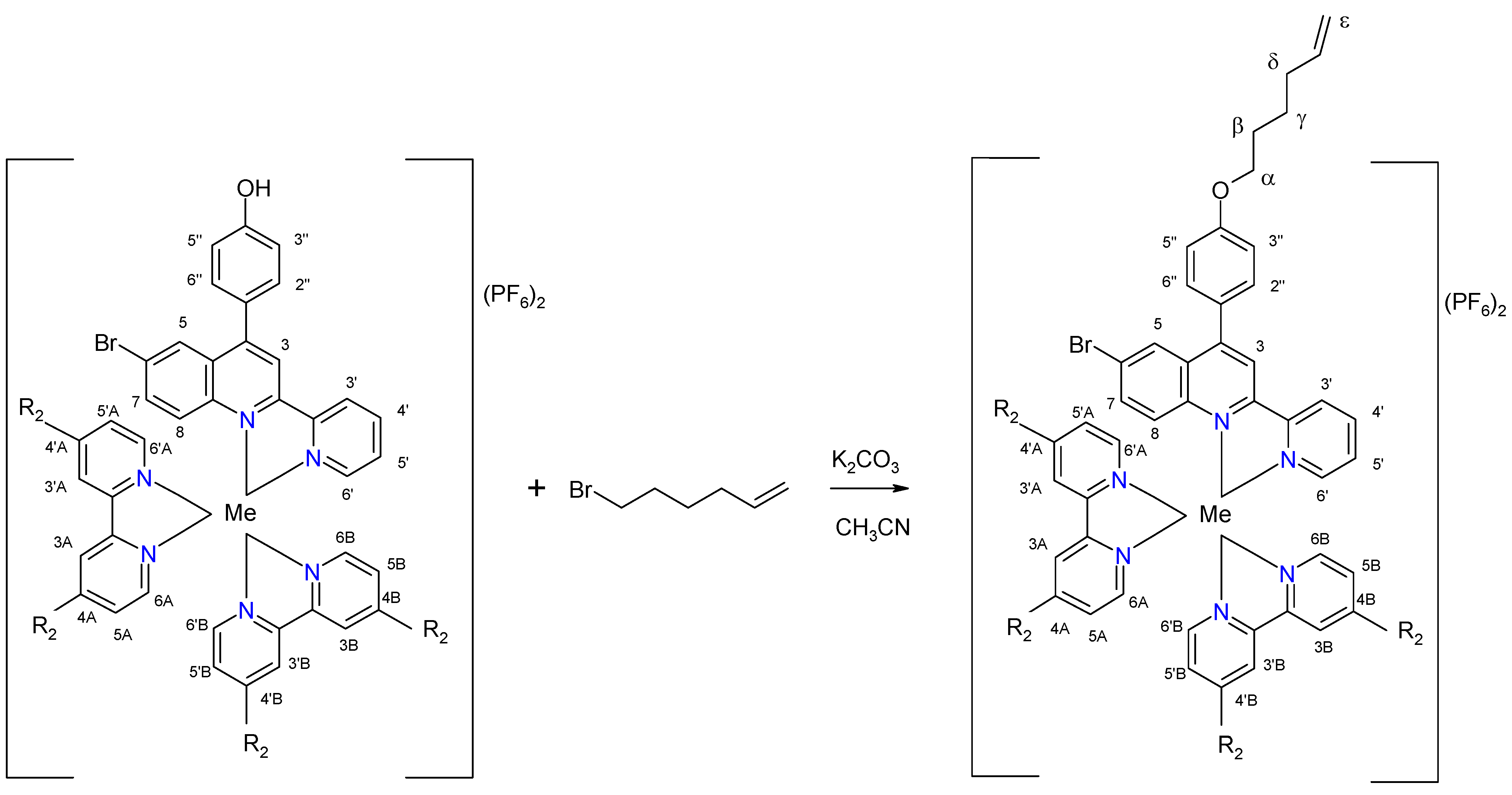
2.2. Complexes
| Complex | Ligand | Me | R1 | R2 | Chemical Formula |
| C1 | L1 | Ru | -OCH3 | H | [Ru(bpy)2L1] (PF6)2 |
| C2 | L2 | Ru | -OH | H | [Ru(bpy)2L2] (PF6)2 |
| C3 | L1 | Os | -OCH3 | H | [Os(bpy)2L1] (PF6)2 |
| C4 | L2 | Os | -OH | H | [Os(bpy)2L2] (PF6)2 |
| C5 | L1 | Ru | -OCH3 | CH3 | [Ru(dmbpy)2L1] (PF6)2 |
| C6 | L2 | Ru | -OH | CH3 | [Ru(dmbpy)2L2] (PF6)2 |
| C7 | L1 | Os | -OCH3 | CH3 | [Os(dmbpy)2L1] (PF6)2 |
| C8 | L2 | Os | -OH | CH3 | [Os(dmbpy)2L2] (PF6)2 |
| Complex | Ligand | Me | R1 | R2 | Chemical Formula |
| C9 | L3 | Ru | O(CH2)4CH=CH2 | H | [Ru(bpy)2L3](PF6)2 |
| C10 | L3 | Os | O(CH2)4CH=CH | H | [Os(bpy)2L3](PF6)2 |
| C11 | L3 | Ru | O(CH2)4CH=CH | CH3 | [Ru(dmbpy)2L3](PF6)2 |
| C12 | L3 | Os | O(CH2)4CH=CH | CH3 | [Os(dmbpy)2L3](PF6)2 |
3. Experimental
3.1. General
3.2. Syntheses
4. Conclusions
Acknowledgements
References and Notes
- Smalley, S.; Waterland, M.; Telfer, S. Heteroleptic dipyrrin/bipyridine complexes of ruthenium(II). Inorg. Chem. 2009, 1, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Bolink, H.; Coronado, E.; Costa, R.; Lardiés, N.; Orti, E. Near-Quantitative internal quantum efficiency in a light-emitting electrochemical cell. Inorg. Chem. 2008, 47, 9149–9151. [Google Scholar] [CrossRef] [PubMed]
- Flamigni, L.; Collin, J.P.; Sauvage, J.P. Iridium terpyridine complexes as functional assembling units in arrays for the conversion of light energy. Acc. Chem. Res. 2008, 41, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Puckett, C.; Barton, J. Mechanism of cellular uptake of a ruthenium polypyridyl complex. Biochemistry 2008, 47, 11711–11716. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, B.; Das, S. Role of new bis(2,2’-bipyridyl)(triazolopyridyl)ruthenium(II) complex in the organic bistable memory application. Chem. Mater. 2008, 20, 1209–1211. [Google Scholar] [CrossRef]
- Zhou, M.; Robertson, G.; Roovers, J. Comparative study of ruthenium(II) tris(bipyridine) derivatives for electrochemiluminescence application. Inorg. Chem. 2005, 44, 8317–8325. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.M. Electrochemiluminescence (ECL). Chem. Rev. 2004, 104, 3003–3036. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chao, H.; Li, H.; Hong, X.; Liu, Y.; Tan, L.; Ji, L. DNA interactions of cobalt(III) mixed-polypyridyl complexes containing asymmetric ligands. J. Inorg. Biochem. 2004, 98, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zou, X.; Liu, J. Shape and enantioselective interaction of Ru(II)/Co(III) polypyridyl complexes with DNA. Coord. Chem. Rev. 2001, 216–217, 513–536. [Google Scholar] [CrossRef]
- Fanni, S.; Keyes, T.; O’Connor, C.; Hughes, H.; Wang, R.; Vos, J. Excited–states properties of ruthenium (II) polypyridyl complexes containing asymmetric triazole ligands. Coord. Chem. Rev. 2000, 208, 77–86. [Google Scholar] [CrossRef]
- Deng, H.; Cai, J.; Xu, H.; Zhang, H.; Ji, L. Ruthenium (II) complexes containing asymmetric ligands: synthesis, characterization, crystal structure and DNA–binding. J. Chem. Soc. Dalton Trans. 2003, 325–330. [Google Scholar] [CrossRef]
- Sortino, S.; Petralia, S.; Conoci, S.; Di Bella, S. Novel self-assembled monolayers of dipolar ruthenium (III/II) pentaamine (4,4’-bipyridinium) complexes on ultrathin platinum films as redox molecular switches. J. Am. Chem. Soc. 2003, 125, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Das, A.; Van der Boom, M.E. Electrochemical addressing of the optical properties of a monolayer on a trasparent conducting substrate. Angew. Chem. Int. Ed. 2005, 44, 3237–3240. [Google Scholar] [CrossRef] [PubMed]
- Mamo, A. Synthesis and characterization of new substituted terdendate 2,6-pyridylquinolines and 1,3-benzenequinoline ligands. J. Heterocyclic. Chem. 2000, 37, 1225–1231. [Google Scholar] [CrossRef]
- Mamo, A.; Stefio, I.; Parisi M., F.; Di Pietro, C.; Campagna, S. Luminescent and redox-active iridium (III)-cyclometallated compounds with terdentate ligands. Inorg. Chem. 1997, 36, 5947–5950. [Google Scholar] [CrossRef] [PubMed]
- Mamo, A.; Stefio, I.; Poggi, A.; Tringali, C.; Di Pietro, C.; Campagna, S. Ruthenium (II) and osmium (II) complexes with new terdentate polyquinoline and cyclometalating ligands. Synthesis, NMR characterization, luminescence properties, and electrochemical behavior. New J. Chem. 1997, 21, 1173–1185. [Google Scholar]
- Mamo, A.; Pappalardo, A. Synthesis and characterization of acyclic and cyclic aza-bridged ligands incorporating 2,2’-bipyridine subunits and their complexes with copper (II), cobalt (II), and nickel (II). Molecules 2003, 8, 521–535. [Google Scholar] [CrossRef]
- Lupo, F.; Fragalà, M.; Gupta, T.; Mamo, A.; Aureliano, A.; Bettinelli, M.; Speghini, A.; Gulino, A. Optical selective CO sensing by a ruthenium monolayer covalently assembled on silica substrates. J. Mat. Chem. 2010, in press. [Google Scholar]
- Campagna, S.; Mamo, A.; Stille, J.K. Synthesis, characterization, luminescence properties, and electrochemical behavior of ruthenium (II) complexes with two new bidentate and tridentate 2-pyridyl-quinoline ligands. J. Chem. Soc. Dalton Trans. 1991, 2545–2551. [Google Scholar] [CrossRef]
- Riego, E.C.; Jin, X.; Thummel, R.P. Introduction of benzo[h]quinoline and 1,10-phenanthroline subunits by Friedlander methodology. J. Org. Chem. 1996, 61, 3017–3022. [Google Scholar]
- Hu, Y.Z.; Zhang, G.; Thummel, R.P. Friedlander approach for the incorporation of 6-bromoquinoline into novel chelating ligands. Org. Lett. 2003, 5, 2251–2253. [Google Scholar] [CrossRef] [PubMed]
- McOmie, J.F.; Watts, M.L.; West, D.E. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron 1968, 24, 2289–2292. [Google Scholar] [CrossRef]
- Lehner, A.; Steinhoff, G.; Brandt, M.; Eickhoff, M.; Stutzmann, M. Hydrosilylation of crystalline silicon (111) and hydrogenated amorphous silicon surfaces: A comparative x-ray photoelectron spectroscopy study. J. Appl. Phys. 2003, 94, 2289–2294. [Google Scholar] [CrossRef]
- Constable, E.C. Homoleptic complexes of 2,2’ bipyridine. Adv. Inorg. Chem. 1989, 34, 1–63. [Google Scholar]
- Lay, P.; Harmer, W.D. Recent advances in osmium chemistry. Adv. Inorg. Chem. 1991, 37, 219–379. [Google Scholar]
- Togano, T.; Nagao, N.; Tsuchida, M.; Kumakura, H.; Hisamatsu, K.; Howell, F.; Mukaida, M. One pot and selective synthesis of a series of [RuCl6-2nLn] (L bidentate ligand n = 0–3) types of complexes with polypyridyl ligands, another example of the synthetic utility of ‘ruthenium-blue’ solution. Inorg. Chim. Acta 1992, 195, 221–225. [Google Scholar] [CrossRef]
Sample Availability: Available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
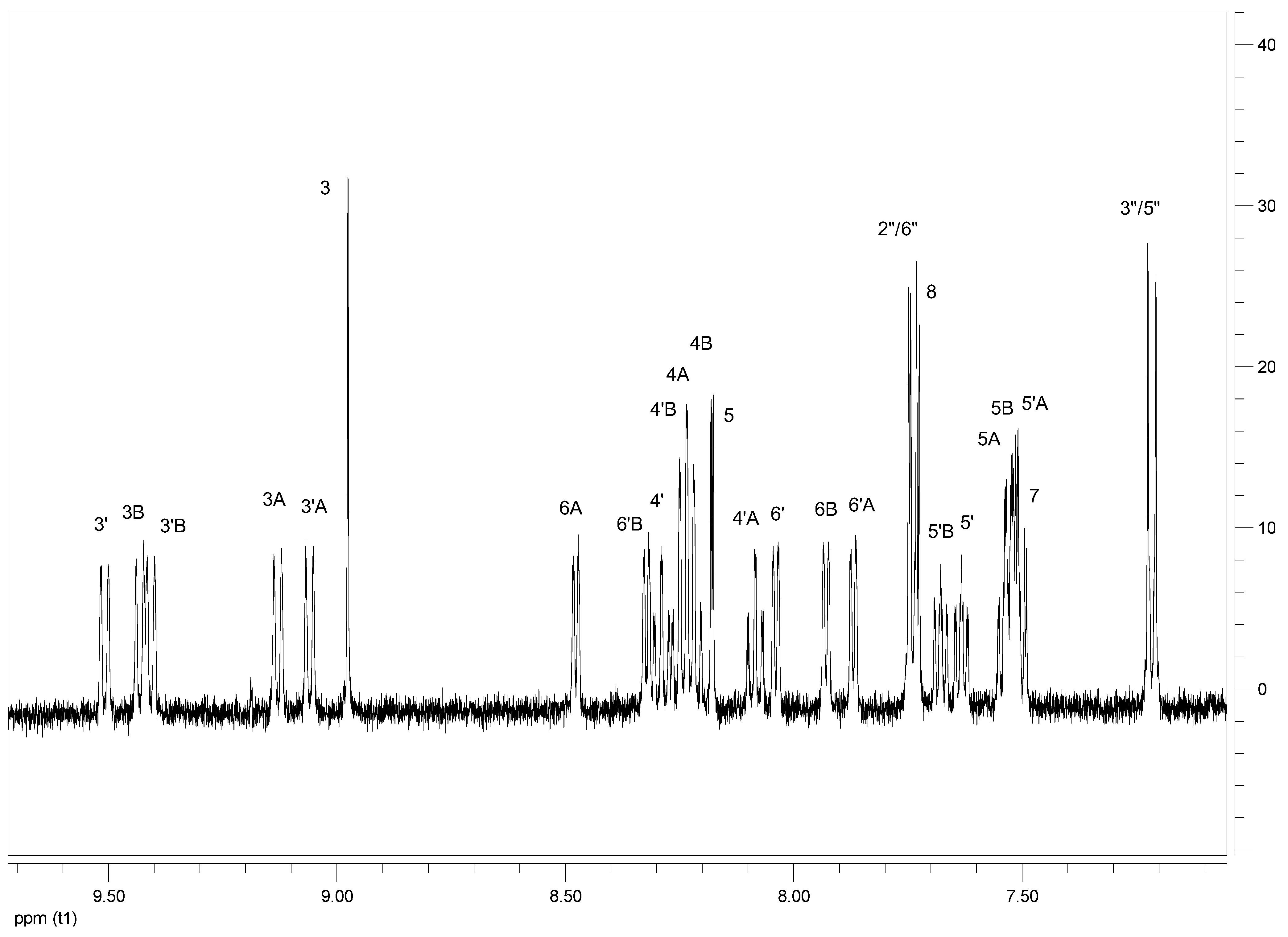
| Proton | 3 | 5 | 7 | 8 | 3′ | 4′ | 5′ | 6′ | 2′′/6′′ | 3′′/5′′ | OCH3 | OH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L1 | 8.51 s | 8.13 d J = 2.0 | 7.80 dd J = 9.0 J = 2.0 | 8.10 d J = 8.0 | 8.60 d J = 8.0 | 7.89 dt J = 8.0 J = 1.5 | 7.38 dt J = 9.0 J = 1.5 | 8.73 d J = 4.0 | 7.52 d J = 9.0 | 7.09 d J = 9.0 | 3.92 s | - |
| L2 | 8.61 s | 8.16 d J = 2.5 | 7.91dd J = 9.0, J = 2.0 | 8.14 d J = 9.0 | 8.73 d J = 8.0 | 8.01 dt J = 8.0, J = 1.5 | 7.50 dt J = 8.5, J = 1.5 | 8.74 d J = 4.0 | 7.53 d J = 9.0 | 7.12 d J = 9.0 | - | 8.79 bs |
| C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C12 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H3 | 8.98s | 8.79s | 8.94s | 8.75s | 9.01s | 8.76s | 8.97s | 8.58s | 8.81s | 8.80s | 8.84s | 8.83s | |
| H5 | 8.18d J = 2.0 | 8.23d J = 2.0 | 8.12d J = 2.0 | 8.17d J = 2.0 | 8.18d J = 2.0 | 8.21d J = 2.0 | 8.12d J = 2.0 | 8.10d J = 2.0 | 8.18d J = 2.5 | 8.15d J = 2.0 | 8.18d J = 2.0 | 8.15dJ = 2.0 | |
| H7 | 7.50 dd J = 9.0, 2.0 | 7.50dd J = 9.0,2.0 | 7.46dd J = 9.0,2.0 | 7.46dd J = 8.5,2.0 | 7.49dd J = 9.0,2.0 | 7.50m | 7.45dd J = 9.0, 2.0 | 7.50m | 7.50dd J = 9.0,2.0 | 7.45dd J = 8.5,2.0 | 7.49dd J = 9.0,2.0 | 7.46dd J = 9.0,2.0 | |
| H8 | 7.74d J = 9.0 | 7.73d J = 9.5 | 7.69d J = 9.0 | 7.68d J = 9.0 | 7.82d J = 9.0 | 8.11d J = 9.0 | 7.77d J = 9.0 | 7.97d J = 9.0 | 7.73d J = 9.5 | 7.67dJ = 9.0 | 8.10d J = 9.5 | 7.84d J = 9.0 | |
| H3′ | 9.51d J = 8.5 | 9.23d J = 9.0 | 9.46d J = 8.5 | 9.16d J = 8.0 | 9.57d J = 9.0 | 9.18d J = 8.5 | 9.52d J = 9.0 | 9.18d J = 8.5 | 9.23d J = 9.0 | 9.36d J = 8.5 | 9.29d J = 8.5 | 9.42d J = 9.0 | |
| H4′ | 8.29 dt J = 8.0, 1.5 | 8.32m | 8.07m | 8.10m | 8.23dt J = 8.0, 1.5 | 8.27dt J = 8.0,2.0 | 8.01m | 8.18m | 8.32dd J = 8.02.0 | 8.07tJ = 8.0 | 8.26t J = 8.0 | 8.01t J = 8.0 | |
| H5′ | 7.63dt J = 7.0,1.5 | 7.65t J = 7.5,1.5 | 7.61dt J = 7.5,1.5 | 7.63m | 7.62dt J = 7.0,1.5 | 7.60t J = 8.0 | 7.60dt J = 7.0,1.5 | 7.62t J = 8.0 | 7.62m | 7.62m | 7.61t J = 7.0 | 7.61t J = 7.0 | |
| H6′ | 8.04d J = 5.0 | 8.06d J = 5.5 | 8.04d J = 5.0 | 8.06d J = 5.0 | 8.02d J = 5.0 | 8.02d J = 5.5 | 8.02d J = 5.0 | 7.96d J = 5.0 | 8.05d J = 5.0 | 7.88dJ = 5.0 | 8.03d J = 5.5 | 7.86d J = 5.0 | |
| H2”/6” | 7.73d J = 9.0 | 7.55d J = 8.5 | 7.72d J = 9.0 | 7.54d J = 8.5 | 7.71d J = 8.5 | 7.55d J = 8.0 | 7.70d J = 8.5 | 7.53d J = 8.5 | 7.70d J = 8.5 | 7.54dJ = 8.5 | 7.68d J = 8.5 | 7.52d J = 8.5 | |
| H3′′/5′′ | 7.22d J = 9.0 | 7.14d J = 8.5 | 7.20d J = 9.0 | 7.12d J = 8.5 | 7.21d J = 8.5 | 7.13d J = 8.0 | 7.19d J = 8.5 | 7.14d J = 8.5 | 7.23d J = 9.0 | 7.15d J = 8.5 | 7.22d J = 8.5 | 7.14d J = 8.5 | |
| H3A | 9.13d J = 8.0 | 8.76d J = 8.0 | 9.08d J = 8.0 | 8.71d J = 8.5 | 9.28bs | 8.62bs | 9.23bs | 8.60bs | 8.77d J = 8.0 | 8.98d J = 8.5 | 8.92bs | 9.13bs | |
| H4A | 8.24m | 8.26m | 8.03m | 8.05m | 8.24dd J = 8.02.0 | 8.05m | |||||||
| H5A | 7.53m | 7.58m | 7.43m | 7.48m | 7.48m | 7.50m | 7.38m | 7.44m | 7.55m | 7.46m | 7.50m | 7.41m | |
| H6A | 8.48d J = 5.0 | 8.52d J = 6.0 | 8.35d J = 5.0 | 8.39d J = 5.5 | 8.22bd | 7.75bd | 8.09m | 7.78m | 8.50d J = 6.0 | 8.38d J = 5.5 | 7.76d J = 5.5 | 7.64d J = 5.0 | |
| H3′A | 9.06d J = 8.0 | 8.68d J = 8.0 | 8.99d J = 8.0 | 8.61d J = 8.0 | 9.19bs | 8.53bs | 9.12bs | 8.48bs | 8.69d J = 9.0 | 8.87d J = 8.0 | 8.83bs | 9.01bs | |
| H4′A | 8.08 dt J = 8.0, 1.0 | 8.11t J = 7.5 | 7.89t J = 8.0 | 7.92t J = 8.0 | 8.11t J = 7.5 | 7.92t J = 8.0 | |||||||
| H5′A | 7.52 m | 7.56m | 7.37 m | 7.41m | 7.31 m | 7.32m | 7.16 m | 7.24 m | 7.52m | 7.44m | 7.31m | 7.23 m | |
| H6′A | 7.87d J = 5.0 | 7.91d J = 6.0 | 7.81d J = 5.5 | 7.85d J = 5.0 | 7.35d J = 5.0 | 7.38d J = 5.5 | 7.29d J = 5.5 | 7.42m | 7.89d J = 5.5 | 7.76d J = 7.5 | 7.37d J = 6.0 | 7.24m | |
| H3B | 9.43d J = 8.0 | 8.96d J = 8.0 | 9.39d J = 8.0 | 8.92bs | 9.61bs | 8,78bs | 9.57bs | 8.74bs | 8.97d J = 8.0 | 9.26d J = 8.0 | 9.15bs | 9.44bs | |
| H4B | 8.23 m | 8.28m | 8.02 m | 8.07m | 8.27 dt J = 8.01.5 | 8.07m | |||||||
| H5B | 7.53 m | 7.57m | 7.30 m | 7.44m | 7.31 m | 7.34m | 7.18 m | 7.30 m | 7.54m | 7.43m | 7.32m | 7.21 m | |
| H6B | 7.93d J = 5.0 | 7.99d J = 5.0 | 7.84d J = 5.0 | 7.90bd | 7.66d J = 5.0 | 7.73d J = 5.0 | 7.67bd | 8.16bd | 7.97d J = 5.5 | 7.85d J = 5.0 | 7.70d J = 6.0 | 7.58d J = 5.5 | |
| H3′B | 9.41d J = 8.0 | 8.93d J = 7.5 | 9.39d J = 7.5 | 8.91bd | 9.60bs | 8.75bs | 9.56bs | 8.70bs | 8.94d J = 8.5 | 9.24bd | 9.13bs | 9.43bs | |
| H4′B | 8.23m | 8.30m | 8.04m | 8.09m | 8.28 dd J = 8.01.5 | 8.09m | |||||||
| H5′B | 7.68dt J = 7.0, 1.5 | 7.70t J = 7.0 | 7.64dt J = 7.0, 1.5 | 7.66m | 7.64d J = 7.0 | 7.65d J = 7.5 | 7.60d J = 7.0 | 7.36m | 7.68t J = 7.5 | 7.66t J = 7.5 | 7.64d J = 7.5 | 7.62d J = 7.0 | |
| H6′B | 8.32d J = 6.0 | 8.36d J = 5.0 | 8.16d J = 6.0 | 8.20d J = 5.5 | 8.22d J = 6.0 | 8.23d J = 6.5 | 8.06d J = 6.0 | 7.70d J = 6.0 | 8.34d J = 5.5 | 8.21d J = 6.5 | 8.20d J = 6.0 | 7.62d J = 6.0 | |
| CH3′A | 2.49bs | 2.45bs | 2.44bs | 2.50bs | 2.48bs | 2.47bs | |||||||
| CH3B | 2.60bs | 2.54bs | 2.53bs | 2.70bs | 2.59bs | 2.53bs | |||||||
| CH3A | 2.60bs | 2.55bs | 2.53bs | 2.71bs | 2.60bs | 2.53bs | |||||||
| CH3′B | 2.60bs | 2.56bs | 2.53bs | 2.72bs | 2.61bs | 2.53bs | |||||||
| αCH2 | 4.17 t J = 6.5 | 4.17 t J = 6.5 | 4.17 t J = 6.5 | 4.17 t J = 6.5 | |||||||||
| βCH2 | 1.87t J = 6.5 | 1.86t J = 6.5 | 1.87t J = 6.5 | 1.87t J = 6.5 | |||||||||
| γCH2 | 1.63t J = 6.5 | 1.62tJ = 6.5 | 1.63t J = 6.5 | 1.63t J = 6.5 | |||||||||
| δCH2 | 2.17dt J = 7.0, 1.5 | 2.15dt J = 7.0, 1.5 | 2.17dt J = 7.0, 1.5 | 2.16dt J = 7.0, 1.5 | |||||||||
| εCH2 | 5.00dd J = 10.0, 2.0 | 5.04dd J = 10.0, 2.0 | 5.02dd J = 10.0, 2.0 | 5.00dd J = 10.0, 2.0 | |||||||||
| -CH | 5.87m | 5.88m | 5.87m | 5.87m | |||||||||
| OCH3 | 3.95s | 3.95s | 3.95s | 3.95s | |||||||||
| -OH | 9.95bs | 9.96bs | 9.95bs | 9.96bs | |||||||||
| Complex. | Yield (%) (XXX) Me2CO/Et2O | Molecular Formula (M.W.) | %C Found (Calcd) | %H Found (Calcd) | %N Found (Calcd) | FAB–MSm/z |
|---|---|---|---|---|---|---|
| C1 | 72 | Ru(C41H31BrF12N6OP2) (1094.50) | 44.96 (45.00) | 3.00 (2.85) | 7.56 (7.67) | 949 [Ru(bpy)2L1](PF6)+ |
| C2 | 63 | Ru(C40H29BrF12N6OP2) (1080.47) | 44.33 (44.46) | 2.85 (2.70) | 7.47 (7.77) | 935 [Ru(bpy)2L2](PF6)+ |
| C3 | 69 | Os(C41H31BrF12N6OP2) (1183.66) | 42.01 (41.62) | 2.22 (2.64) | 7.37 (7.10) | 1038 [Os(bpy)2L1](PF6)+ |
| C4 | 71 | Os(C40H29BrF12N6OP2) (1169.63) | 41.74 (41.41) | 2.09 (2.50) | 7.33 (7.18) | 1024 [Os(bpy)2L2](PF6)+ |
| C5 | 95 | Ru(C45H39BrF12N6OP2) (1150.60) | 46.91 (46.97) | 3.57 (3.40) | 6.94 (7.30) | 1005 [Ru(dmbpy)2L1](PF6)+ |
| C6 | 80 | Ru(C44H37BrF12N6OP2) (1136.57) | 46.11 (46.49) | 3.19 (3.28) | 7.41 (7.39) | 991 [Ru(dmbpy)2L2](PF6)+ |
| C7 | 73 | Os(C45H39BrF12N6OP2) (1239.76) | 46.52 (46.13) | 2.49 (2.90) | 6.74 (6.78) | 1094 [Os(dmbpy)2L1](PF6)+ |
| C8 | 80 | Os(C44H37BrF12N6OP2) (1225.73) | 42.98 (43.11) | 2.85 (3.04) | 6.92 (6.89) | 1080 [Os(dmbpy)2L2](PF6)+ |
| C9 | 99 | Ru(C46H39BrF12N6OP2) (1162.61) | 47.63 (47.52) | 3.49 (3.38) | 6.93 (7.23) | 1017 [Ru(bpy)2L3](PF6)+ |
| C10 | 94 | Os(C46H39BrF12N6OP2) (1251.77) | 44.52 (44.13) | 3.06 (3.14) | 6.58 (6.71) | 1106 [Ru(bpy)2L3](PF6)+ |
| C11 | 83 | Ru(C50H47BrF12N6OP2) (1218.65) | 48.91 (49.27) | 4.11 (3.89) | 6.87 (6.89) | 1099 [Ru(dmbpy)2L3](PF6)+ |
| C12 | 88 | Os(C50H47BrF12N6OP2) (1307.81) | 46.09 (45.91) | 3.36 (3.62) | 6.80 (6.42) | 1188 [Os(dmbpy)2L3](PF6)+ |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mamo, A.; Aureliano, A.; Recca, A. Asymmetric Ruthenium(II) and Osmium(II) Complexes with New Bidentate Polyquinoline Ligands. Synthesis and NMR Characterization. Molecules 2010, 15, 1324-1339. https://doi.org/10.3390/molecules15031324
Mamo A, Aureliano A, Recca A. Asymmetric Ruthenium(II) and Osmium(II) Complexes with New Bidentate Polyquinoline Ligands. Synthesis and NMR Characterization. Molecules. 2010; 15(3):1324-1339. https://doi.org/10.3390/molecules15031324
Chicago/Turabian StyleMamo, Antonino, Alessandro Aureliano, and Antonino Recca. 2010. "Asymmetric Ruthenium(II) and Osmium(II) Complexes with New Bidentate Polyquinoline Ligands. Synthesis and NMR Characterization" Molecules 15, no. 3: 1324-1339. https://doi.org/10.3390/molecules15031324
APA StyleMamo, A., Aureliano, A., & Recca, A. (2010). Asymmetric Ruthenium(II) and Osmium(II) Complexes with New Bidentate Polyquinoline Ligands. Synthesis and NMR Characterization. Molecules, 15(3), 1324-1339. https://doi.org/10.3390/molecules15031324