Automation in the High-throughput Selection of Random Combinatorial Libraries—Different Approaches for Select Applications

Abstract
:Introduction

Selection of Random Combinatorial Libraries


{kind=link}
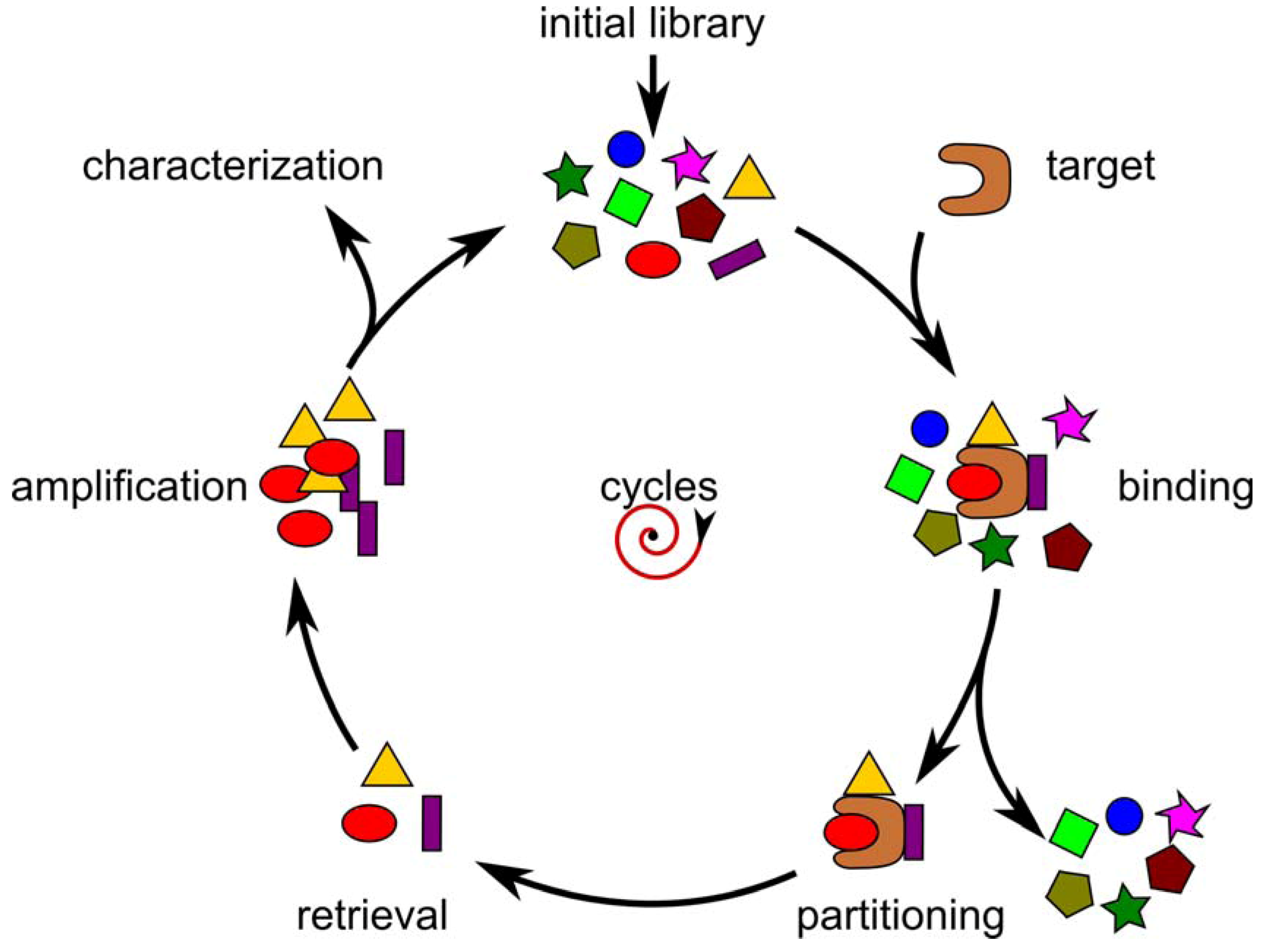{kind=link}
{kind=link}
| Technology | Phenotype | Genotype | Link | Diversities | Reference | Amplification/synthesis |
|---|---|---|---|---|---|---|
| Phage display | peptide/protein | ssDNA | viral particle | 106–1010 | [16] | in vivo/bacteria |
| Bacterial display | peptide/protein | plasmid | intracellular | 108–1011 | [17] | in vivo/bacteria |
| Yeast display | peptide/protein | plasmid | intracellular | 109 | [18] | in vivo/yeast |
| Ribosome display | peptide/protein | mRNA | complexed | 1013 | [19] | in vitro/cell free expression |
| mRNA display | peptide/protein | mRNA | covalent | 1013 | [20] | in vitro/cell free expression |
| in vitro compartimentalisation | protein | DNA | micelle compartment | 108–1011 | [21] | in vitro/cell free expression |
| RNA SELEX | RNA | RNA | covalent | 1015 | [22] | in vitro |
| DNA SELEX | DNA | DNA | covalent | 1015 | [22] | in vitro |
| PNA display | PNA | DNA | colavent | 108 | [14] | in vitro/chemical |
| DNA display | synthetic compound | DNA | covalent | 108 | [23] | in vitro/chemical |
Applications and Choice of Selection Technology
| Binder | Peptide | Antibody | RNA | DNA | Spiegelmer |
|---|---|---|---|---|---|
| Biological stability | medium | strong | low | medium | strong |
| Chemical stability | strong | low-medium | medium | strong | as RNA or DNA |
| M ultiple regeneration | yes | no | yes | yes | yes |
| Synthesis | chemical/in vivo | cell culture | chemical/in vivo | chemical | chemical |
| Adverse immune reactions | no (size and structure dependent) | yes (needs humanisation) | no | no | no |
| Synthesis cost | low-medium | high | low-medium | low | medium-high |
| Selectivity/affinity | low-medium | high | high | medium-high | as RNA or DNA |
| Phage display | RNA SELEX | DNA SELEX | |
|---|---|---|---|
| Target selection | Incubation, partitioning, retrieval, 2 hours | Incubation, partitioning, retrieval, 2 hours | Incubation, partitioning, retrieval, 2 hours |
| Amplification | reinfection, growth, superinfection, purification, 1–2 days | reverse transcription, PCR, transcription, purification, 2 days | PCR, ssDNA generation, purification, 4–6 hours |
| Selection cycles | 4 | 10 to 15 | 10 to 15 |
| Duration of selection | 5–8 days | 20–45 days | 10–20 days |
| Cloning | reinfection, colony generation, picking, glycerol stocks, 2 days | vector ligation, transformation, colony generation, picking, glycerol stocks, 2 days | vector ligation, transformation, colony generation, picking, glycerol stocks, 2 days |
| Characterization | growth, superinfection, phage ELISA, 2 days | PCR, transcription, purification, FLAA, 2 days | PCR, ssDNA preparation, purification, FLAA, 2 days |
| Total duration | 9–12 days | 24–49 days | 14–24 days |
| Total cost of consumables | low | medium-high | low |
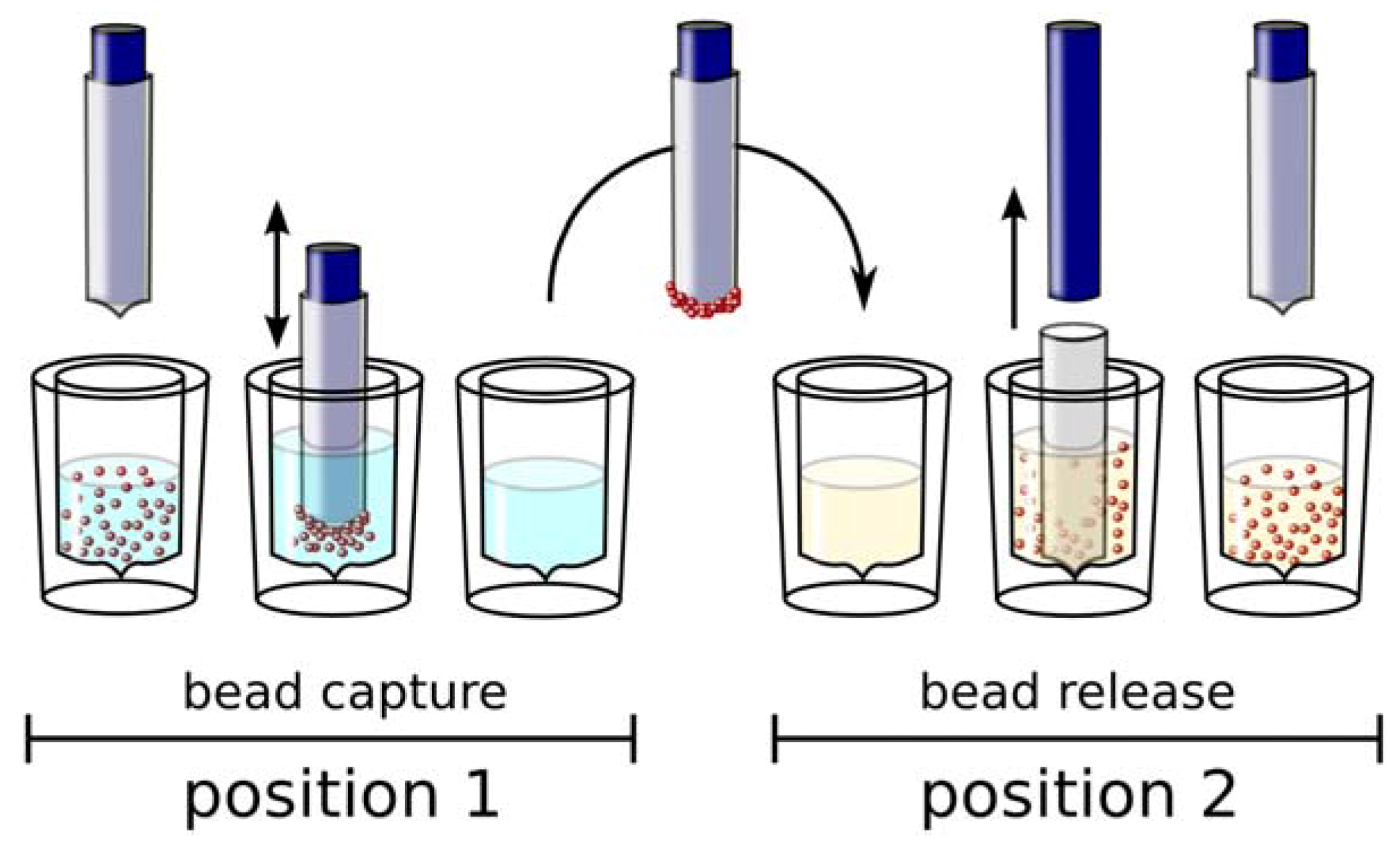
Automation of Selection and Monitoring

Conclusions and Outlook
Acknowledgements
- Sample Availability: Not available.
References
- Parmley, S.F.; Smith, G.P. Filamentous fusion cloning vectors for the study of epitopes and design of vaccines. Adv. Exp. Med. Biol. 1989, 251, 215–218. [Google Scholar]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random peptide libraries: A source of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar]
- Breitling, F.; Dübel, S.; Seehaus, T.; Klewinghaus, I.; Little, M. A surface expression vector for antibody screening. Gene 1991, 104, 147–153. [Google Scholar] [CrossRef]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Konthur, Z.; Crameri, R. High-throughput applications of phage display in proteomic analyses. TARGETS 2003, 2, 261–270. [Google Scholar] [CrossRef]
- Mead, D.A.; Kemper, B. Chimeric single-stranded DNA phage-plasmid cloning vectors. Biotechnology (Reading, Mass.) 1988, 10, 85–102. [Google Scholar]
- Houshmand, H.; Fröman, G.; Magnusson, G. Use of bacteriophage T7 displayed peptides for determination of monoclonal antibody specificity and biosensor analysis of the binding reaction. Anal. Biochem. 1999, 268, 363–370. [Google Scholar]
- Maruyama, I.N.; Maruyama, H.I.; Brenner, S. Lambda foo: A lambda phage vector for the expression of foreign proteins. Proc. Nat. Acad. Sci. USA 1994, 91, 8273–8277. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Leemhuis, H.; Stein, V.; Griffiths, A.D.; Hollfelder, F. New genotype-phenotype linkages for directed evolution of functional proteins. Curr. Opin. Struct. Biol. 2005, 15, 472–478. [Google Scholar] [CrossRef]
- Brudno, Y.; Birnbaum, M.E.; Kleiner, R.E.; Liu, D.R. An in vitro translation, selection and amplification system for peptide nucleic acids. Nat. Chem. Biol. 2010, 6, 148–155. [Google Scholar]
- Wrenn, S.J.; Harbury, P.B. Chemical evolution as a tool for molecular discovery. Ann. Rev. Biochem. 2007, 76, 331–349. [Google Scholar] [CrossRef]
- Rothe, C.; Urlinger, S.; Löhning, C.; Prassler, J.; Stark, Y.; Jäger, U.; Hubner, B.; Bardroff, M.; Pradel, I.; Boss, M.; Bittlingmaier, R.; Bataa, T.; Frisch, C.; Brocks, B.; Honegger, A.; Urban, M. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J. Mol. Biol. 2008, 376, 1182–1200. [Google Scholar] [CrossRef]
- Daugherty, P.S. Protein engineering with bacterial display. Curr. Opin. Struct. Biol. 2007, 17, 474–480. [Google Scholar] [CrossRef]
- Feldhaus, M.J.; Siegel, R.W.; Opresko, L.K.; Coleman, J.R.; Feldhaus, J.M.W.; Yeung, Y.A.; Cochran, J.R.; Heinzelman, P.; Colby, D.; Swers, J.; Graff, C.; Wiley, H.S.; Wittrup, K.D. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat. Biotechnol. 2003, 21, 163–170. [Google Scholar]
- He, M.; Taussig, M.J. Eukaryotic ribosome display with in situ DNA recovery. Nat. Methods 2007, 4, 281–288. [Google Scholar] [CrossRef]
- Takahashi, T.T.; Roberts, R.W. In vitro selection of protein and peptide libraries using mRNA display. Meth. Mol. B. 2009, 535, 293–314. [Google Scholar] [CrossRef]
- Miller, O.J.; Bernath, K.; Agresti, J.J.; Amitai, G.; Kelly, B.T.; Mastrobattista, E.; Taly, V.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Directed evolution by in vitro compartmentalization. Nat. Methods 2006, 3, 561–570. [Google Scholar] [CrossRef]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX--a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef]
- Wrenn, S.J.; Weisinger, R.M.; Halpin, D.R.; Harbury, P.B. Synthetic ligands discovered by in vitro selection. J. Amer. Chem. Soc. 2007, 129, 13137–13143. [Google Scholar] [CrossRef]
- Ulrich, H.; Trujillo, C.A.; Nery, A.A.; Alves, J.M.; Majumder, P.; Resende, R.R.; Martins, A.H. DNA and RNA aptamers: From tools for basic research towards therapeutic applications. Comb. Chem. High Throughput Screen. 2006, 9, 619–632. [Google Scholar] [CrossRef]
- Thie, H.; Meyer, T.; Schirrmann, T.; Hust, M.; Dübel, S. Phage display derived therapeutic antibodies. Curr. Pharm. Biotechnol. 2008, 9, 439–446. [Google Scholar] [CrossRef]
- Famulok, M.; Hartig, J.S.; Mayer, G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem. Rev. 2007, 107, 3715–3743. [Google Scholar] [CrossRef]
- Almagro, J.C.; Fransson, J. Humanization of antibodies. Front. Biosci. 2008, 13, 1619–1633. [Google Scholar]
- Bugaut, A.; Toulmé, J.-J.; Rayner, B. SELEX and dynamic combinatorial chemistry interplay for the selection of conjugated RNA aptamers. Org. Biomol. Chem. 2006, 4, 4082–4088. [Google Scholar] [CrossRef]
- Kimoto, M.; Endo, M.; Mitsui, T.; Okuni, T.; Hirao, I.; Yokoyama, S. Site-specific incorporation of a photo-crosslinking component into RNA by T7 transcription mediated by unnatural base pairs. Chem. Biol. 2004, 11, 47–55. [Google Scholar]
- Adler, A.; Forster, N.; Homann, M.; Göringer, H.U. Post-SELEX chemical optimization of a trypanosome-specific RNA aptamer. Comb. Chem. High Throughput Scr. 2001, 4, 193–205. [Google Scholar]
- Vater, A.; Klussmann, S. Toward third-generation aptamers: Spiegelmers and their therapeutic prospects. Curr. Opin. Drug Discov. Dev. 2003, 6, 253–261. [Google Scholar]
- Hutanu, D.; Remcho, V.T. Aptamers as molecular recognition elements in chromatographic separations. Adv. Chromatogr. 2007, 45, 173–196. [Google Scholar] [CrossRef]
- Peyrin, E. Nucleic acid aptamer molecular recognition principles and application in liquid chromatography and capillary electrophoresis. J. Sep. Sci. 2009, 32, 1531–1536. [Google Scholar] [CrossRef]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gústafsdóttir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar] [CrossRef]
- Tombelli, S.; Mascini, M. Aptamers as molecular tools for bioanalytical methods. Curr. Opin. Mol. Ther. 2009, 11, 179–188. [Google Scholar]
- Hust, M.; Toleikis, L.; Dübel, S. Selection Strategies II: Antibody Phage Display. In Handbook of Therapeutic Antibodies; Dübel, S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp. 45–68. [Google Scholar]
- Hall, B.; Micheletti, J.M.; Satya, P.; Ogle, K.; Pollard, J.; Ellington, A.D. Design, synthesis, and amplification of DNA pools for in vitro selection. Curr. Protoc. Nucleic Acid Chem. 2009. unit 9.2. [Google Scholar]
- Stanlis, K.K.H.; McIntosh, J.R. Single-strand DNA aptamers as probes for protein localization in cells. J. Histochem. Cytochem. 2003, 51, 797–808. [Google Scholar] [CrossRef]
- Hamilton, S. Introduction to screening automation. Meth. Mol. B. 2003, 190, 169–193. [Google Scholar]
- Cohen, S.; Trinka, R.F. Fully automated screening systems. Meth. Mol. B. 2002, 190, 213–228. [Google Scholar]
- Menke, K.C. Unit automation in high throughput screening. Meth. Mol. B. 2002, 190, 195–212. [Google Scholar]
- Konthur, Z. Automation of Selection and Engineering. In Handbook of Therapeutic Antibodies; Dübel, S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp. 412–430. [Google Scholar]
- Koehn, J.; Hunt, I. High-Throughput Protein Production (HTPP): a review of enabling technologies to expite protein production. Meth. Mol. B. 2009, 498, 1–18. [Google Scholar] [CrossRef]
- Huber, R.; Ritter, D.; Hering, T.; Hillmer, A.-K.; Kensy, F.; Müller, C.; Wang, L.; Büchs, J. Robo-Lector - a novel platform for automated high-throughput cultivations in microtiter plates with high information content. Microb. Cell Fact. 2009, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.C.; Ellington, A.D. Automated selection of anti-protein aptamers. Bioorg. Med. Chem. 2001, 9, 2525–2531. [Google Scholar] [CrossRef]
- Cox, J.C.; Rudolph, P.; Ellington, A.D. Automated RNA selection. Biotechnol. Progr. 1998, 14, 845–850. [Google Scholar] [CrossRef]
- Eulberg, D.; Buchner, K.; Maasch, C.; Klussmann, S. Development of an automated in vitro selection protocol to obtain RNA-based aptamers: Identification of a biostable substance P antagonist. Nucl. Acid. Res. 2005, 33, e45. [Google Scholar] [CrossRef]
- Hybarger, G.; Bynum, J.; Williams, R.F.; Valdes, J.J.; Chambers, J.P. A microfluidic SELEX prototype. Anal. Bioanal. Chem. 2006, 384, 191–198. [Google Scholar] [CrossRef]
- Krebs, B.; Rauchenberger, R.; Reiffert, S.; Rothe, C.; Tesar, M.; Thomassen, E.; Cao, M.; Dreier, T.; Fischer, D.; Höss, A.; Inge, L.; Knappik, A.; Marget, M.; Pack, P.; Meng, X.Q.; Schier, R.; Söhlemann, P.; Winter, J.; Wölle, J.; Kretzschmar, T. High-throughput generation and engineering of recombinant human antibodies. J. Immunol. Methods 2001, 254, 67–84. [Google Scholar] [CrossRef]
- McConnell, S.J.; Dinh, T.; Le, M.H.; Spinella, D.G. Biopanning phage display libraries using magnetic beads vs. polystyrene plates. BioTechniques 1999, 26, 208–210, 214. [Google Scholar]
- Cox, J.C.; Hayhurst, A.; Hesselberth, J.; Bayer, T.S.; Georgiou, G.; Ellington, A.D. Automated selection of aptamers against protein targets translated in vitro: From gene to aptamer. Nucl. Acid. Res. 2002, 30, e108. [Google Scholar] [CrossRef]
- Walter, G.; Konthur, Z.; Lehrach, H. High-throughput screening of surface displayed gene products. Comb. Chem. High Throughput Screen. 2001, 4, 193–205. [Google Scholar]
- Konthur, Z.; Walter, G. Automation of phage display for high-throughput antibody development. Targets 2002, 1, 30–36. [Google Scholar] [CrossRef]
- Wochner, A.; Cech, B.; Menger, M.; Erdmann, V.A.; Glökler, J. Semi-automated selection of DNA aptamers using magnetic particle handling. BioTechniques 2007, 43. 344, 346, 348 passim. [Google Scholar]
- Wochner, A.; Glökler, J. Nonradioactive fluorescence microtiter plate assay monitoring aptamer selections. BioTechniques 2007, 42. 578, 580, 582. [Google Scholar]
- Williams, S.C.; Badley, R.A.; Davis, P.J.; Puijk, W.C.; Meloen, R.H. Identification of epitopes within beta lactoglobulin recognised by polyclonal antibodies using phage display and PEPSCAN. J. Immunol. Methods 1998, 213, 1–17. [Google Scholar] [CrossRef]
- Wochner, A.; Menger, M.; Orgel, D.; Cech, B.; Rimmele, M.; Erdmann, V.A.; Glökler, J. A DNA aptamer with high affinity and specificity for therapeutic anthracyclines. Anal. Biochem. 2008, 373, 34–42. [Google Scholar]
- Schütze, T.; Arndt, P.F.; Menger, M.; Wochner, A.; Vingron, M.; Erdmann, V.A.; Lehrach, H.; Kaps, C.; Glökler, J. A calibrated diversity assay for nucleic acid libraries using DiStRO--a Diversity Standard of Random Oligonucleotides. Nucl. Acid. Res. 2009. gkp1108. [Google Scholar]
- Baum, P.D.; McCune, J.M. Direct measurement of T-cell receptor repertoire diversity with AmpliCot. Nat. Methods 2006, 3, 895–901. [Google Scholar] [CrossRef]
- Britten, R.J.; Kohne, D.E. Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science 1968, 161, 529–540. [Google Scholar]
- Weigand, J.E.; Sanchez, M.; Gunnesch, E.; Zeiher, S.; Schroeder, R.; Suess, B. Screening for engineered neomycin riboswitches that control translation initiation. RNA (New York, NY) 2008, 14, 89–97. [Google Scholar]
- Hallborn, J.; Carlsson, R. Automated screening procedure for high-throughput generation of antibody fragments. BioTechniques 2002, Suppl., 30–37. [Google Scholar]
- Turunen, L.; Takkinen, K.; Söderlund, H.; Pulli, T. Automated panning and screening procedure on microplates for antibody generation from phage display libraries. J. Biomol. Screen. 2009, 14, 282–293. [Google Scholar] [CrossRef]
- Buckler, D.R.; Park, A.; Viswanathan, M.; Hoet, R.M.; Ladner, R.C. Screening isolates from antibody phage-display libraries. Drug Discov. Today 2008, 13, 318–324. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Glökler, J.; Schütze, T.; Konthur, Z. Automation in the High-throughput Selection of Random Combinatorial Libraries—Different Approaches for Select Applications. Molecules 2010, 15, 2478-2490. https://doi.org/10.3390/molecules15042478
Glökler J, Schütze T, Konthur Z. Automation in the High-throughput Selection of Random Combinatorial Libraries—Different Approaches for Select Applications. Molecules. 2010; 15(4):2478-2490. https://doi.org/10.3390/molecules15042478
Chicago/Turabian StyleGlökler, Jörn, Tatjana Schütze, and Zoltán Konthur. 2010. "Automation in the High-throughput Selection of Random Combinatorial Libraries—Different Approaches for Select Applications" Molecules 15, no. 4: 2478-2490. https://doi.org/10.3390/molecules15042478
APA StyleGlökler, J., Schütze, T., & Konthur, Z. (2010). Automation in the High-throughput Selection of Random Combinatorial Libraries—Different Approaches for Select Applications. Molecules, 15(4), 2478-2490. https://doi.org/10.3390/molecules15042478