Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches

Abstract
:1. Introduction
2. Results and Discussion
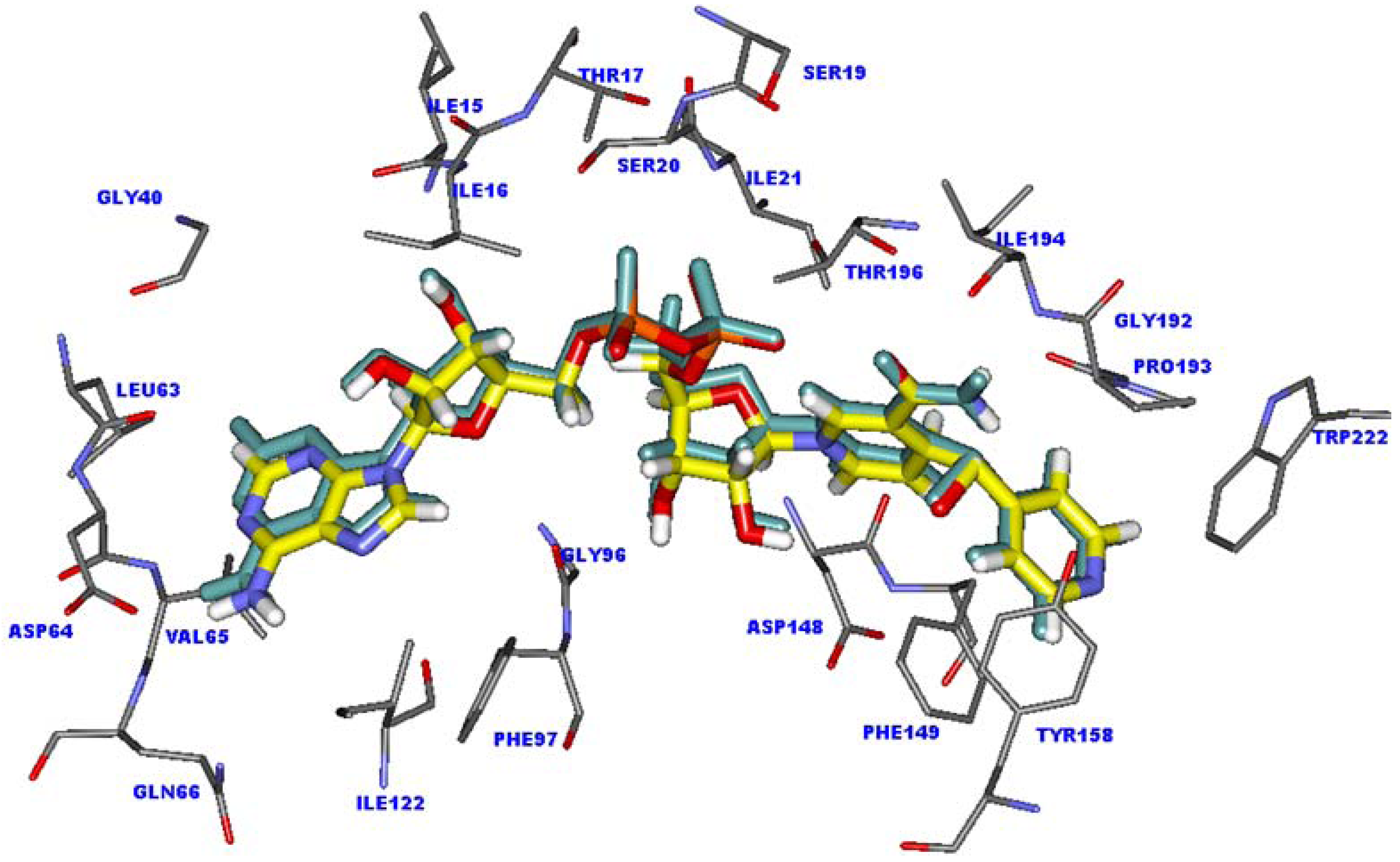
2.1. Validation of the molecular docking calculations
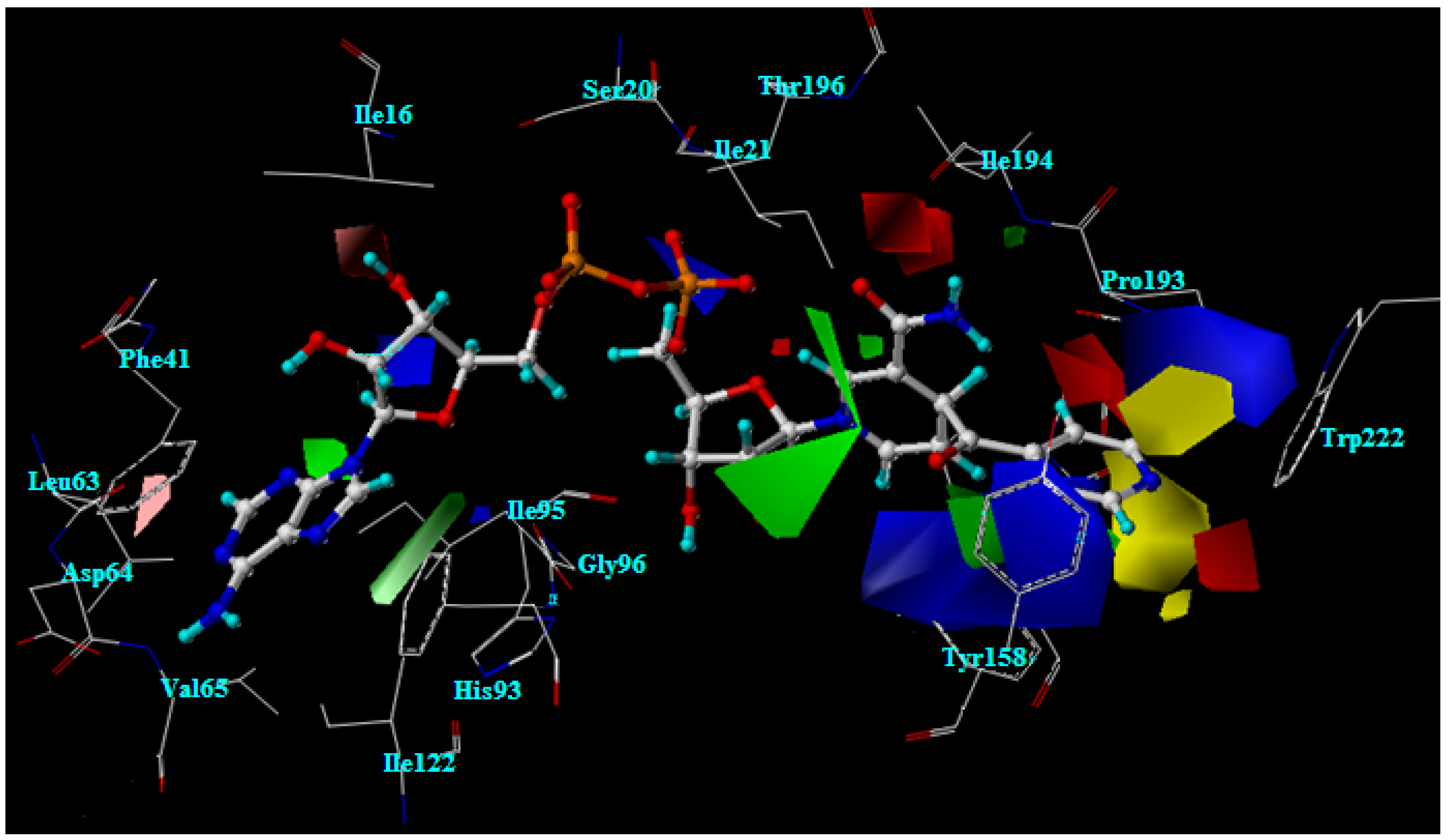
2.2. Molecular docking analysis of the highly active compounds


| Compound | R | Log (1/MIC) | Compound | R | Log (1/MIC) |
|---|---|---|---|---|---|
| INH |  | 7.70 | 18 |  | 5.10 |
| 1 |  | 7.22 | 19 [a] |  | 5.10 |
| 2[a] |  | 6.82 | 20 |  | 4.92 |
| 3 |  | 6.52 | 21[a] |  | 4.92 |
| 4 |  | 6.40 | 22 |  | 4.92 |
| 5 |  | 6.22 | 23 |  | 4.82 |
| 6 |  | 6.10 | 24 |  | 4.70 |
| 7 |  | 5.82 | 25 |  | 4.52 |
| 8 |  | 5.70 | 26[a] |  | 4.52 |
| 9[a] |  | 5.70 | 27 |  | 4.40 |
| 10[a] |  | 5.52 | 28 |  | 4.10 |
| 11 |  | 5.52 | 29 |  | 4.00 |
| 12 |  | 5.52 | 30 |  | 4.00 |
| 13 |  | 5.52 | 31 |  | 3.65 |
| 14 |  | 5.22 | 32 |  | 3.52 |
| 15 |  | 5.22 | 33 |  | 3.22 |
| 16 |  | 5.22 | 34 |  | 3.22 |
| 17 |  | 5.10 | 35 |  | 3.22 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd. | Nicotinamide | Pyrophosphate | Adenine ring | |||
|---|---|---|---|---|---|---|
| Asp148 | Thr196 | Ser20 | Ile21 | Asp64 | Val65 | |
| INH | 2.75, 2.34 | 2.99, 3.66 | 2.47 | 2.69 | 2.09 | 2.80 |
| 1 | 2.74, 2.33 | 2.92, 3.65 | 2.42 | 2.63 | 2.14 | 2.82 |
| 2 | 2.91, 2.40 | 2.97, 3.66 | 2.43 | 2.72 | 2.12 | 2.74 |
| 3 | 2.80, 2.36 | 2.85, 3.59 | 2.42 | 2.59 | 2.21 | 2.86 |
| 4 | 2.87, 2.36 | 2.94, 3.64 | 2.43 | 2.68 | 2.16 | 2.76 |
| 5 | 2.91, 2.41 | 2.89, 3.62 | 2.41 | 2.66 | 2.25 | 2.76 |
| 6 | 2.79, 2.35 | 2.91, 3.65 | 2.40 | 2.62 | 2.13 | 2.81 |
| 7 | 2.87, 2.34 | 2.91, 3.68 | 2.41 | 2.64 | 2.23 | 2.75 |
| 8 | 2.95, 2.39 | 2.90, 3.61 | 2.43 | 2.67 | 2.25 | 2.76 |
| 9 | 3.71, 3.38 | 3.21, 4.53 | 2.13 | 3.22 | 1.64 | 2.45 |
| 10 | 2.93, 2.99 | 3.31, 4.38 | 2.28 | 3.14 | 1.56 | 2.43 |
| 11 | 3.29, 2.81 | 3.10, 3.77 | 2.30 | 2.90 | 1.78 | 2.65 |
| 12 | 3.01, 2.50 | 2.91, 3.68 | 2.41 | 2.73 | 2.16 | 2.71 |
| 13 | 2.87, 2.34 | 2.90, 3.67 | 2.41 | 2.63 | 2.22 | 2.76 |
| 14 | 3.46, 3.15 | 3.12, 3.88 | 2.14 | 2.97 | 1.67 | 2.60 |
| 15 | 3.20, 2.85 | 2.87, 3.74 | 2.29 | 2.82 | 1.88 | 2.55 |
| 16 | 3.09, 2.53 | 3.12, 3.84 | 2.39 | 2.86 | 1.89 | 2.58 |
| 17 | 2.67, 2.99 | 3.31, 4.53 | 2.28 | 3.14 | 1.56 | 2.43 |
| 18 | 3.83, 3.62 | 3.30, 4.71 | 2.11 | 3.34 | 1.72 | 2.59 |
| 19 | 2.82, 2.36 | 3.02, 3.64 | 2.83 | 2.73 | 2.35 | 2.86 |
| 20 | 3.38, 2.89 | 3.08, 3.83 | 2.21 | 2.89 | 1.77 | 2.59 |
| 21 | 3.28, 2.56 | 3.10, 3.67 | 2.30 | 2.90 | 1.78 | 2.65 |
| 22 | 3.38, 3.03 | 2.82, 3.76 | 2.18 | 2.82 | 1.81 | 2.58 |
| 23 | 3.55, 3.51 | 3.43, 4.66 | 2.19 | 3.37 | 1.53 | 2.60 |
| 24 | 2.85, 2.33 | 2.91, 3.67 | 2.41 | 2.64 | 2.23 | 2.75 |
| 25 | 2.72, 2.39 | 2.89, 3.70 | 2.41 | 2.66 | 2.25 | 2.76 |
| 26 | 3.25, 2.81 | 2.90, 3.70 | 2.38 | 2.88 | 1.92 | 2.55 |
| 27 | 2.78, 2.95 | 2.89, 3.66 | 3.01 | 3.15 | 2.15 | 2.61 |
| 28 | 3.06, 2.77 | 3.42, 5.17 | 2.15 | 3.07 | 2.62 | 2.26 |
| 29 | 2.35, 2.24 | 3.02, 3.66 | 2.91 | 2.70 | 2.50 | 2.93 |
| 30 | 2.34, 2.23 | 3.02, 3.56 | 2.83 | 2.73 | 2.35 | 2.86 |
| 31 | 4.66, 3.79 | 3.17, 4.46 | 2.32 | 3.35 | 2.15 | 2.69 |
| 32 | 2.25, 2.38 | 3.05, 3.68 | 2.70 | 2.71 | 2.09 | 2.78 |
| 33 | 3.90, 3.07 | 2.73, 3.57 | 2.16 | 2.81 | 2.02 | 2.68 |
| 34 | 3.52, 3.16 | 3.45, 5.03 | 2.19 | 3.19 | 1.96 | 2.59 |
| 35 | 2.93, 2.42 | 2.99, 3.68 | 2.42 | 2.74 | 2.01 | 2.75 |

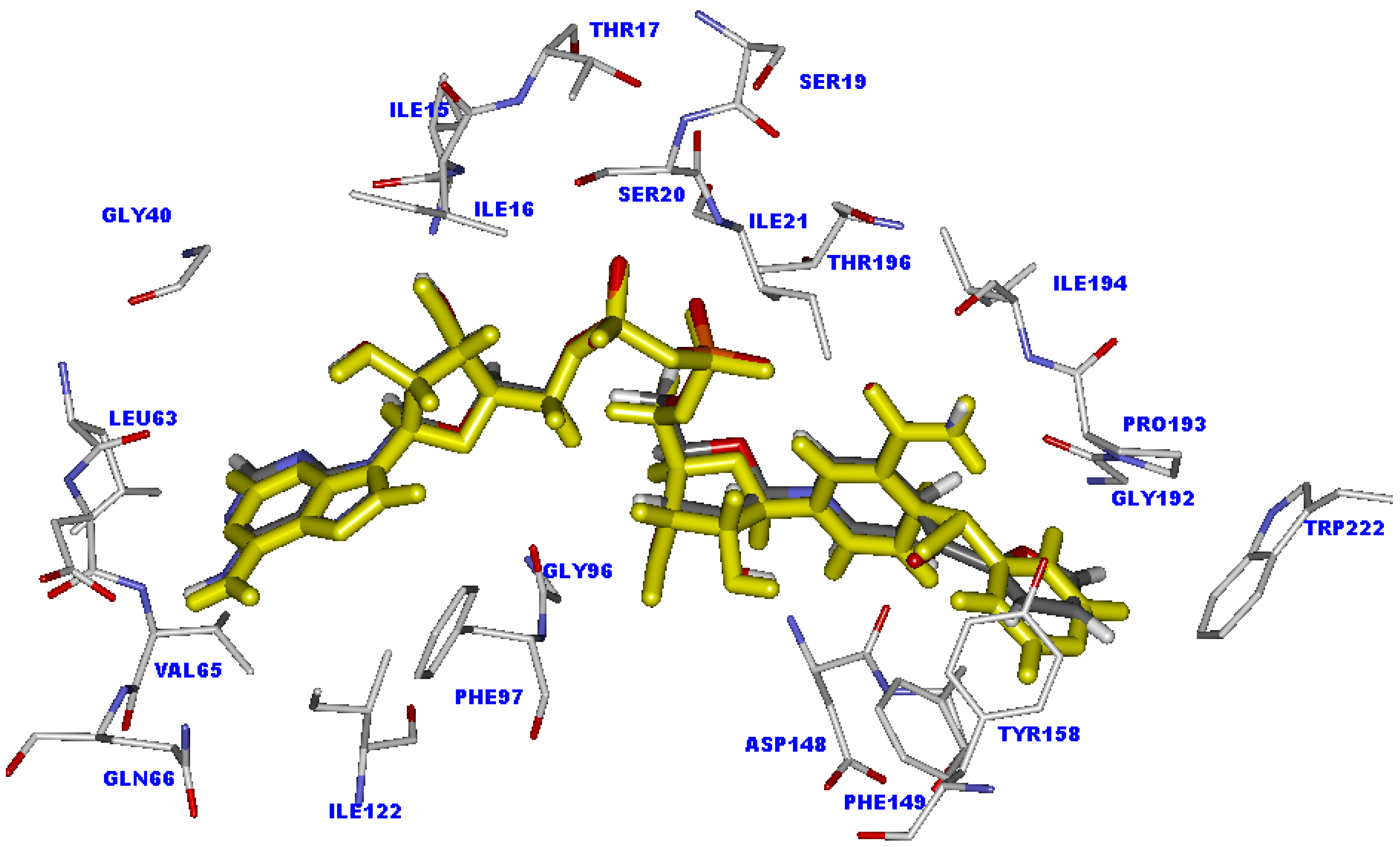
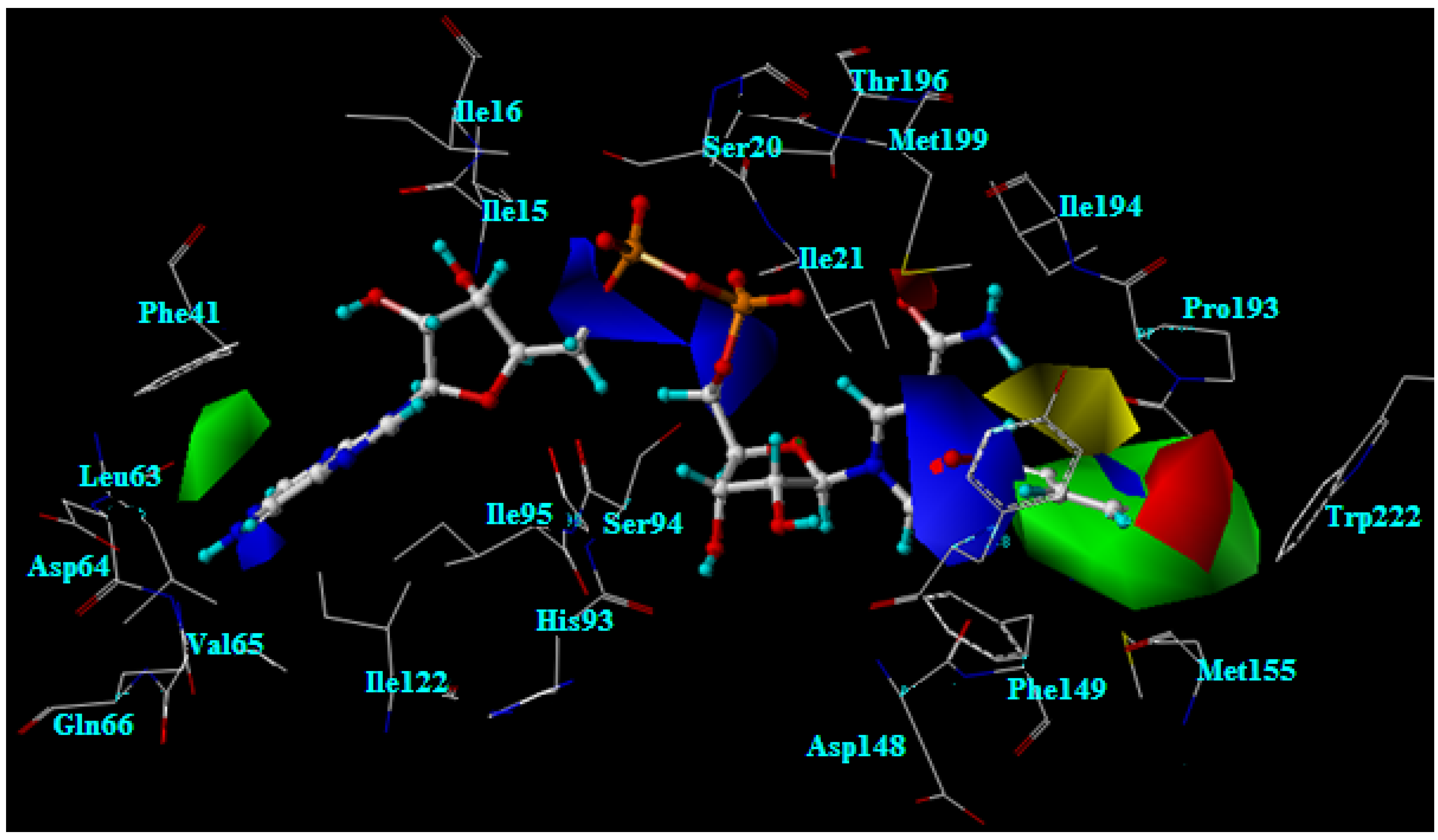
2.3. Docking analysis of the moderately active compounds
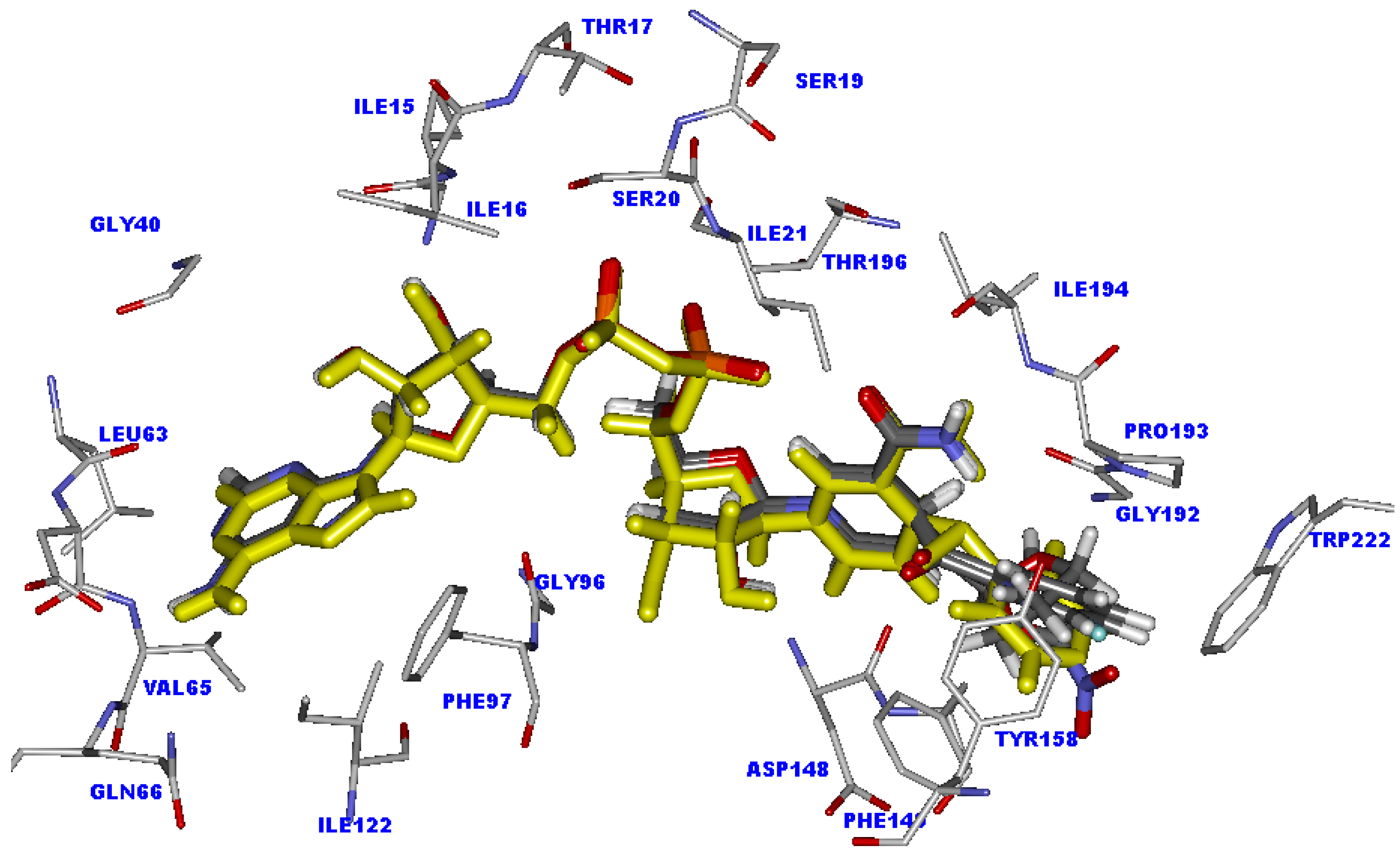
2.4. Docking analysis of the weakly active compounds

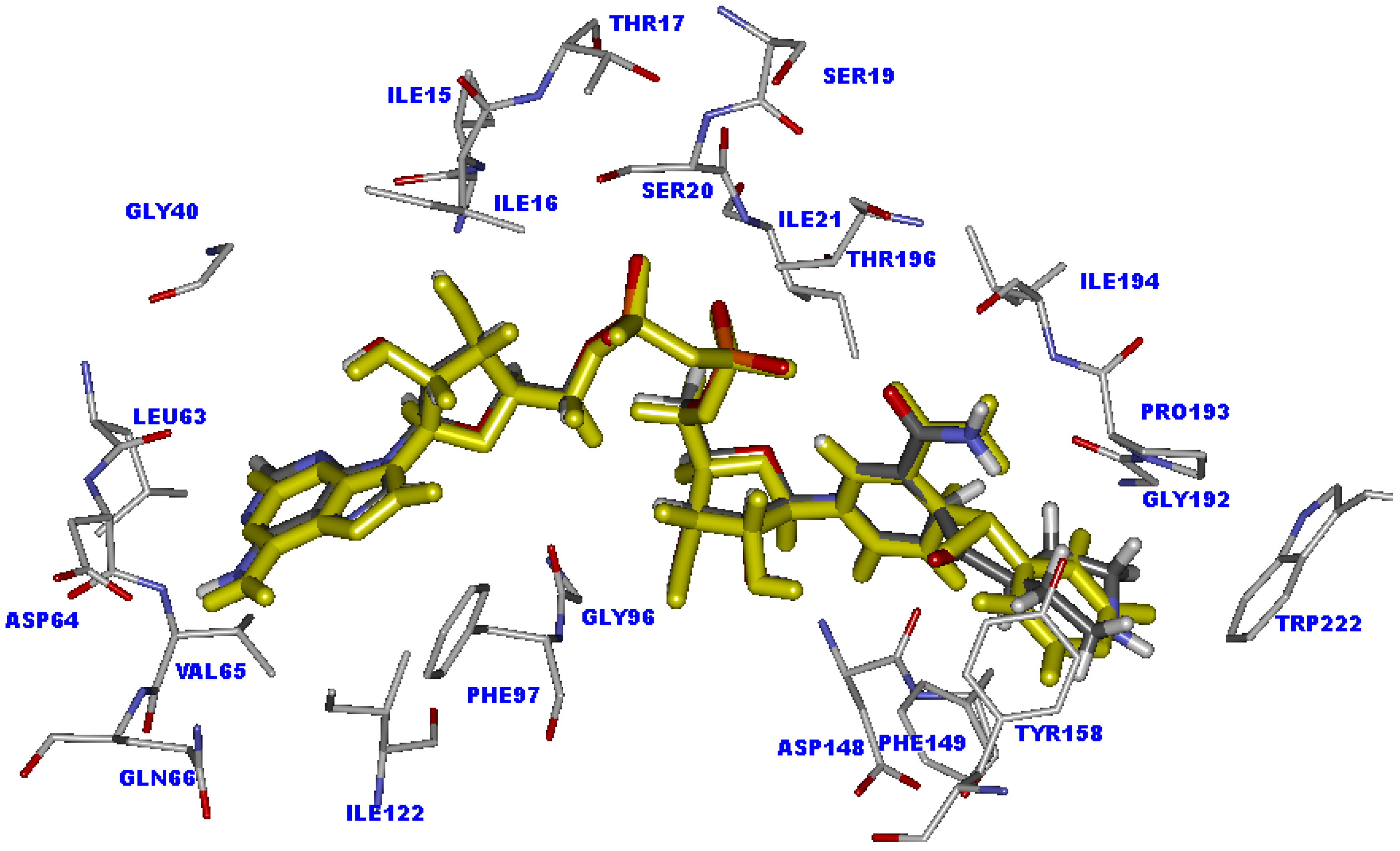
2.5. The favorable interactions for binding of INH derivative adducts
2.6. Interaction energy of the INH-NAD adduct in the InhA binding pocket
2.7. Interaction energy of the highly active compounds in the InhA binding pocket
| Adduct fragment | Amino acid | Interaction energy (kcal/mol) | |||
|---|---|---|---|---|---|
| INH | Cpd. 1 | Cpd. 33 | Cpd. 35 | ||
| R substituent | Phe149 | -1.21 | -0.93 | 30.17 | 2.64 |
| Tyr158 | -2.09 | -0.98 | 23.02 | -0.07 | |
| Ala191 | 0.78 | 3.69 | 18.01 | 4.01 | |
| Gly192 | 1.61 | 1.96 | -2.92 | 5.13 | |
| Pro193 | 0.23 | -0.89 | 23.68 | 6.72 | |
| Trp222 | 0.03 | -1.48 | 18.17 | -0.21 | |
| Nicotinamide | Ile21 | -0.62 | 0.54 | -0.61 | -0.39 |
| Met147 | -0.58 | -0.23 | -0.08 | -0.32 | |
| Asp148 | -4.31 | -5.98 | -4.65 | -5.75 | |
| Phe149 | 6.95 | 2.50 | -0.20 | 4.55 | |
| Lys165 | 1.56 | 1.90 | 3.43 | 2.64 | |
| Ala191 | -2.41 | -1.84 | -1.10 | -1.90 | |
| Gly192 | -0.12 | -0.57 | -0.24 | -0.35 | |
| Pro193 | 0.52 | 0.62 | -0.71 | -0.22 | |
| Ile194 | 13.20 | 10.70 | 3.34 | 3.74 | |
| Thr196 | -1.88 | -1.88 | -2.66 | -2.09 | |
| Nicotinamide Ribose | Gly14 | -0.18 | -0.21 | -0.21 | -0.20 |
| Ser20 | 0.01 | 0.01 | -0.02 | 0.01 | |
| Ile21 | -0.54 | 0.29 | 0.39 | -0.19 | |
| Ala22 | -0.02 | -0.04 | -0.04 | -0.03 | |
| Ser94 | -1.30 | -0.41 | -1.71 | -1.15 | |
| Ile95 | 0.22 | -0.32 | 0.00 | -0.17 | |
| Gly96 | -1.19 | -1.08 | -1.21 | -1.24 | |
| Met147 | 0.54 | 3.76 | -1.27 | 0.99 | |
| Asp148 | -0.29 | -0.61 | 0.26 | -0.39 | |
| Phe149 | -0.26 | -0.23 | -0.22 | -0.24 | |
| Met161 | -0.18 | -0.16 | -0.18 | -0.16 | |
| Lys165 | -11.15 | -11.87 | -10.19 | -12.00 | |
| Ala191 | -0.30 | -0.31 | -0.27 | -0.30 | |
| Pyrophosphate | Gly14 | 8.87 | 9.29 | 9.79 | 9.34 |
| Ile16 | -10.17 | -10.28 | -5.55 | -10.12 | |
| Thr17 | -7.32 | -7.81 | -9.80 | -7.83 | |
| Ser19 | -1.09 | -1.24 | -1.77 | -1.24 | |
| Ser20 | -33.01 | -32.55 | -13.41 | -32.08 | |
| Ile21 | -23.92 | -22.51 | -23.39 | -23.87 | |
| Ala22 | -10.86 | -11.12 | -10.26 | -10.84 | |
| Ser94 | 6.63 | 9.15 | 7.41 | 8.70 | |
| Ile95 | -4.14 | -4.10 | -4.19 | -4.16 | |
| Gly96 | 3.27 | 3.35 | 3.41 | 3.41 | |
| Met147 | -3.63 | -3.57 | -2.99 | -3.49 | |
| Thr196 | -24.48 | -24.93 | -25.04 | -24.94 | |
| Adenine Ribose | Gly14 | 2.62 | 6.28 | 6.74 | 6.42 |
| Ile15 | -1.36 | -1.32 | -1.32 | -1.37 | |
| Ile16 | 3.17 | 3.92 | 2.92 | 2.96 | |
| Ser20 | -0.09 | -0.03 | -0.02 | -0.04 | |
| Phe41 | 0.10 | 0.27 | 3.00 | 0.65 | |
| Val65 | 0.04 | 0.06 | 0.07 | 0.06 | |
| Ser94 | -0.15 | 0.17 | -0.10 | 0.07 | |
| Ile95 | -2.08 | -1.55 | -1.88 | -1.64 | |
| Gly96 | -1.74 | -1.79 | -1.36 | -1.70 | |
| Phe97 | -0.50 | -0.36 | -0.35 | -0.36 | |
| Ile122 | 0.11 | 0.12 | 0.11 | 0.12 | |
| Adenine | Gly14 | -0.65 | -0.80 | -0.71 | -0.76 |
| Gly40 | -1.36 | -0.20 | -0.13 | -0.18 | |
| Phe41 | -2.03 | -2.48 | -2.35 | -2.45 | |
| Leu63 | 0.18 | 1.32 | 1.51 | 1.67 | |
| Asp64 | -9.11 | -8.43 | -7.54 | -8.80 | |
| Val65 | -2.67 | 4.18 | 14.11 | 5.76 | |
| Gln66 | 0.85 | 1.31 | 1.66 | 1.45 | |
| Ile95 | -0.95 | 0.45 | 0.90 | 0.15 | |
| Gly96 | -1.31 | -1.18 | -0.88 | -1.08 | |
| Phe97 | -0.03 | 0.05 | 0.04 | 0.05 | |
| Ile122 | -0.94 | -0.30 | -0.93 | -0.70 | |
2.8. Interaction energy of the weakly active compounds in the InhA binding pocket
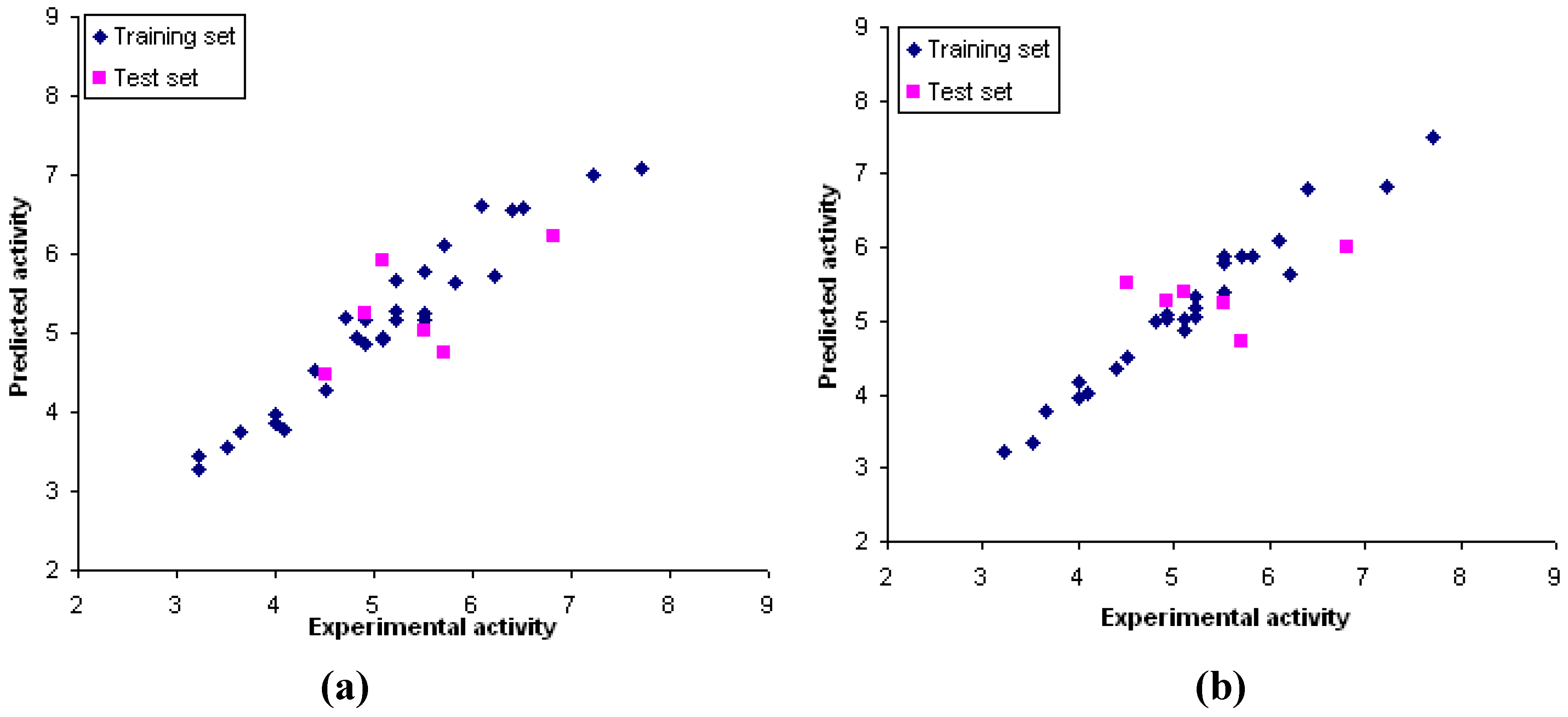
2.9. CoMFA and CoMSIA models
| Models | Statistical parameters | Fraction | |||||
|---|---|---|---|---|---|---|---|
| r2cv | r2 | N | s-press | SEE | F | ||
| CoMFA | 0.67 | 0.94 | 5 | 0.71 | 0.31 | 72.74 | 91/9 (S/E) |
| CoMSIA | |||||||
| S/E | 0.74 | 0.96 | 6 | 0.64 | 0.25 | 82.78 | 43/57 |
| S/E/H | 0.55 | 0.87 | 5 | 0.86 | 0.46 | 28.30 | 17.4/21.2/61.4 |
| S/E/HD | 0.56 | 0.93 | 6 | 0.86 | 0.35 | 42.94 | 27.1/28.6/44.3 |
| S/E/HA | 0.62 | 0.93 | 6 | 0.77 | 0.34 | 41.26 | 32.2/38.9/28.9 |
| S/E/HD/HA | 0.38 | 0.95 | 5 | 1.14 | 0.28 | 86.84 | 18.2/22.9/39.3/19.6 |
| /E/H/HD/HA | 0.32 | 0.91 | 5 | 1.06 | 0.40 | 39.84 | 11.1/13.4/40.5/26.3/8.7 |

2.10. CoMFA and CoMSIA contour analysis of INH derivative adducts


2.11. The steric contour analysis of INH derivative adducts
2.12. The electrostatic contour analysis of INH derivative adducts
2.13. The structural requirement of the R substituent of INH derivative adducts
3. Experimental
3.1. Biological activity data
3.2. Geometry optimization
3.3. Molecular docking calculations
3.4. Interaction energy calculations
3.5. Training and test sets
3.6. Molecular alignment rules for CoMFA and CoMSIA modeling
4. Conclusions
Acknowledgements
References and Notes
- World Health Organization, Global Tuberculosis Control: Surveillance, Planning, Financing; WHO: Geneva, Switzerland, 2008.
- Ducati, R.G.; Ruffino-Netto, A.; Basso, L.A.; Santos, D.S. The resumption of consumption-A review on tuberculosis. Mem. Inst. Oswaldo Cruz 2006, 101, 697–714. [Google Scholar]
- Aoyagi, T. Chemotherapy for tuberculosis; yesterday, today, and tomorrow--basic and clinical studies. Kekkaku 1998, 73, 459–470. [Google Scholar]
- Paramasivan, C.N.; Herbert, D.; Umapathy, K.C.; Rahman, F.; Krishnamurthy, P.V.; Prabhakar, R. Early bactericidal action of pulsed exposure to rifampicin, ethambutol, isoniazid & pyrazinamide in pulmonary tuberculosis patients. Indian J. Med Res. 1994, 100, 1–4. [Google Scholar]
- Dutt, A.K.; Stead, W.W. Tuberculosis chemotherapy today. Ann. Acad. Med. Singapore 1985, 14, 508–514. [Google Scholar]
- Centers for Disease Control and Prevention (CDC). Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs—worldwide, 2000-2004. MMWR. Morb. Mortal. Wkly. Rep. 2006, 55, 301–305.
- Bloom, B.R.; Murray, C.J. Tuberculosis: Commentary on a reemergent killer. Science 1992, 257, 1055–1064. [Google Scholar]
- Chakaya, J.; Getahun, H.; Granich, R.; Havlir, D. Confronting TB/HIV in the era of increasing anti-TB drug resistance. J. Int. AIDS Soc. 2008, 11, 6. [Google Scholar] [CrossRef]
- El-Sadr, W.M.; Tsiouris, S.J. HIV-associated tuberculosis: Diagnostic and treatment challenges. Semin. Respir. Crit. Care Med. 2008, 29, 525–531. [Google Scholar] [CrossRef]
- Nunn, P.; Reid, A.; De Cock, K.M. Tuberculosis and HIV infection: the global setting. J. Infect. Dis. 2007, 196, S5–S14. [Google Scholar] [CrossRef]
- Morehead, R.S. Delayed death from pulmonary tuberculosis: Unsuspected subtherapeutic drug levels. S. Med. J. 2000, 93, 507–510. [Google Scholar]
- Wheeler, P.R.; Anderson, P.M. Determination of the primary target for isoniazid in mycobacterial mycolic acid biosynthesis with Mycobacterium aurum A+. Biochem. J. 1996, 318, 451–457. [Google Scholar]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R., Jr. InhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar]
- Marrakchi, H.; Lanéelle, G.; QuéMard, A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 2000, 146, 289–296. [Google Scholar]
- Dessen, A.; Quémard, A.; Blanchard, J.S.; Jacobs, W.R., Jr.; Sacchettini, J.C. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 1995, 267, 1638–1641. [Google Scholar]
- Quémard, A.; Sacchettini, J.C.; Dessen, A.; Vilcheze, C.; Bittman, R.; Jacobs, W.R., Jr.; Blanchard, J.S. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 1995, 34, 8235–8241. [Google Scholar]
- Rozwarski, D.A; Grant, G.A.; Barton, D.H.; Jacobs, Jr., W.R.; Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 1998, 279, 98–102. [Google Scholar]
- Lei, B.; Wei, C.J.; Tu, S.C. Action mechanism of antitubercular isoniazid. Activation by Mycobacterium tuberculosis KatG, isolation, and characterization of inha inhibitor. J. Biol. Chem. 2000, 275, 2520–2526. [Google Scholar]
- Saint-Joanis, B.; Souchon, H.; Wilming, M.; Johnsson, K.; Alzari, P.M.; Cole, S.T. Use of site-directed mutagenesis to probe the structure, function and isoniazid activation of the catalase/peroxidase, KatG, from Mycobacterium tuberculosis. Biochem. J. 1999, 338, 753–760. [Google Scholar] [CrossRef]
- Zhao, X; Yu, H.; Yu, S.; Wang, F.; Sacchettini, J.C.; Magliozzo, R.S. Hydrogen peroxide-mediated isoniazid activation catalyzed by Mycobacterium tuberculosis catalase-peroxidase (KatG) and its S315T mutant. Biochemistry 2006, 45, 4131–4140. [Google Scholar]
- Johnsson, K.; King, D.S.; Schultz, P.G. Studies on the mechanism of action of isoniazid and ethionamide in the chemotherapy of tuberculosis. J. Am. Chem. Soc. 1995, 117, 5009–5010. [Google Scholar]
- Metcalfe, C.; Macdonald, I.K.; Murphy, E.J.; Brown, K.A.; Raven, E.L.; Moody, P.C. The tuberculosis prodrug isoniazid bound to activating peroxidases. J. Biol. Chem. 2008, 283, 6193–6200. [Google Scholar]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 62, 1220–1227. [Google Scholar] [CrossRef]
- Nguyen, M.; Claparols, C.; Bernadou, J.; Meunier, B. A fast and efficient metal-mediated oxidation of isoniazid and identification of isoniazid-NAD(H) adducts. Chem. Bio. Chem. 2001, 2, 877–883. [Google Scholar]
- Rawat, R.A.; Whitty, P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 13881–13886. [Google Scholar]
- Dias, M.V.; Vasconcelos, I.B.; Prado, A.M.; Fadel, V.; Basso, L.A.; de Azevedo, W.F., Jr.; Santos, D.S. Crystallographic studies on the binding of isonicotinyl-NAD adduct to wild-type and isoniazid resistant 2-trans-enoyl-ACP (CoA) reductase from Mycobacterium tuberculosis. J. Struct. Biol. 2007, 159, 369–380. [Google Scholar] [CrossRef]
- Oliveira, J.S.; Pereira, J.H.; Canduri, F.; Rodrigues, N.C.; de Souza, O.N.; de Azevedo, W.F., Jr.; Basso, L.A.; Santos, D.S. Crystallographic and pre-steady-state kinetics studies on binding of NADH to wild-type and isoniazid-resistant enoyl-ACP(CoA) reductase enzymes from Mycobacterium tuberculosis. J. Mol. Biol. 2006, 359, 646–666. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar]
- The Global Alliance for TB Drug Development (TB Alliance). Handbook of Anti-Tuberculosis Agents. Tuberculosis 2008, 88, 112–116. [CrossRef]
- Barry, C.E.; Slayden, R.A., 3rd; Mdluli, K. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis. Drug Resist. Updat. 1998, 1, 128–134. [Google Scholar] [CrossRef]
- Larsen, M.H.; Vilchèze, C.; Kremer, L.; Besra, G.S.; Parsons, L.; Salfinger, M.; Heifets, L.; Hazbon, M.H.; Alland, D.; Sacchettini, J.C.; Jacobs, W.R., Jr. Overexpression of inhA, but not kasA, confers resistance to isoniazid and ethionamide in Mycobacterium smegmatis, M. bovis BCG and M. tuberculosis. Mol. Microbiol. 2002, 46, 453–466. [Google Scholar] [CrossRef]
- Wahab, H.A.; Choong, Y.S.; Ibrahim, P.; Sadikun, A.; Scior, T. Elucidating isoniazid resistance using molecular modeling. J. Chem. Inf. Model. 2009, 49, 97–107. [Google Scholar] [CrossRef]
- Pasqualoto, K.F.; Ferreira, E.I.; Santos-Filho, O.A.; Hopfinger, A.J. Rational design of new antituberculosis agents: Receptor-independent four-dimensional quantitative structure-activity relationship analysis of a set of isoniazid derivatives. J. Med. Chem. 2004, 47, 3755–3764. [Google Scholar] [CrossRef]
- Bagchi, M.C.; Maity, B.C.; Bose, S. QSAR of anti tuberculosis drugs of INH type using graphical invariants. J. Mol. Struc. Theochem. 2004, 679, 179–186. [Google Scholar] [CrossRef]
- Andrade, C.H.; Salum Lde, B.; Castilho, M.S.; Pasqualoto, K.F.; Ferreira, E.I.; Andricopulo, A.D. Fragment-based and classical quantitative structure-activity relationships for a series of hydrazides as antituberculosis agents. Mol. Divers. 2008, 12, 47–59. [Google Scholar]
- Ventura, C.; Martins, F. Application of quantitative structure-activity relationships to the modeling of antitubercular compounds. 1. The hydrazide family. J. Med. Chem. 2008, 51, 612–624. [Google Scholar] [CrossRef]
- Cramer, III, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar]
- Klebe, G; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar]
- Klopman, G.; Fercu, D.; Jacob, J. Computer-aided study of the relationship between structure and antituberculosis activity of a series of isoniazid derivatives. Chem. Phys. 1996, 204, 181–193. [Google Scholar] [CrossRef]
- GaussView 03, Revision 3.07; Gaussian, Inc.: Wallingford, CT, USA, 2006.
- Morris, G.M.; Goodshell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Punkvang, A.; Saparpakorn, P.; Hannongbua, S.; Wolschann, P.; Pungpo, P. Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches. Molecules 2010, 15, 2791-2813. https://doi.org/10.3390/molecules15042791
Punkvang A, Saparpakorn P, Hannongbua S, Wolschann P, Pungpo P. Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches. Molecules. 2010; 15(4):2791-2813. https://doi.org/10.3390/molecules15042791
Chicago/Turabian StylePunkvang, Auradee, Patchreenart Saparpakorn, Supa Hannongbua, Peter Wolschann, and Pornpan Pungpo. 2010. "Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches" Molecules 15, no. 4: 2791-2813. https://doi.org/10.3390/molecules15042791
APA StylePunkvang, A., Saparpakorn, P., Hannongbua, S., Wolschann, P., & Pungpo, P. (2010). Elucidating Drug-Enzyme Interactions and Their Structural Basis for Improving the Affinity and Potency of Isoniazid and Its Derivatives Based on Computer Modeling Approaches. Molecules, 15(4), 2791-2813. https://doi.org/10.3390/molecules15042791