Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins

Abstract
:1. Introduction
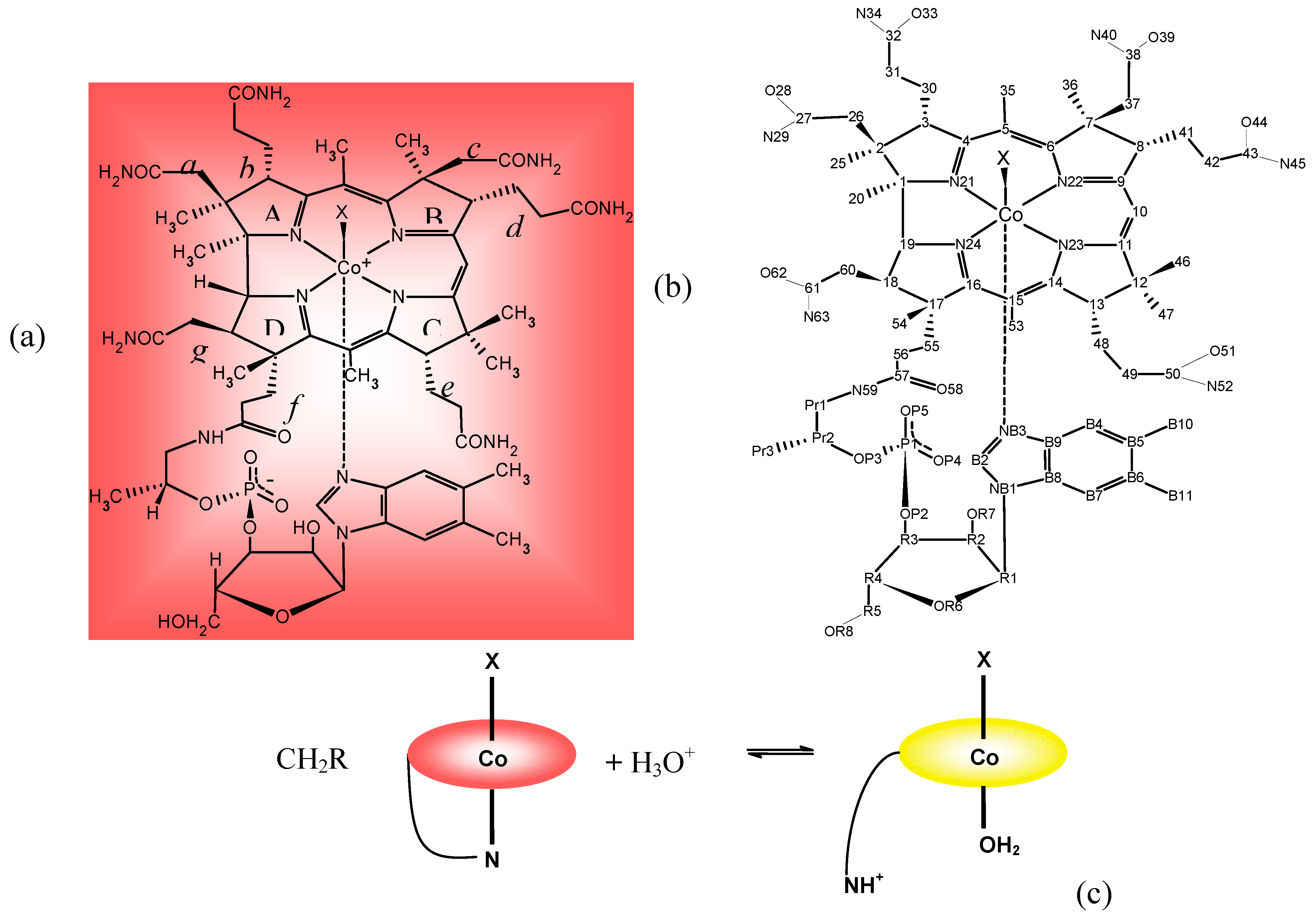
2. The B12 Cofactors
| 1) | RCo(III)alamin → cob(II)alamin + R− |
| 2) | RCo(III)alamin → cob(III)alamin + R− |
| 3) | RCo(III)alamin → cob(I)alamin + R+ |
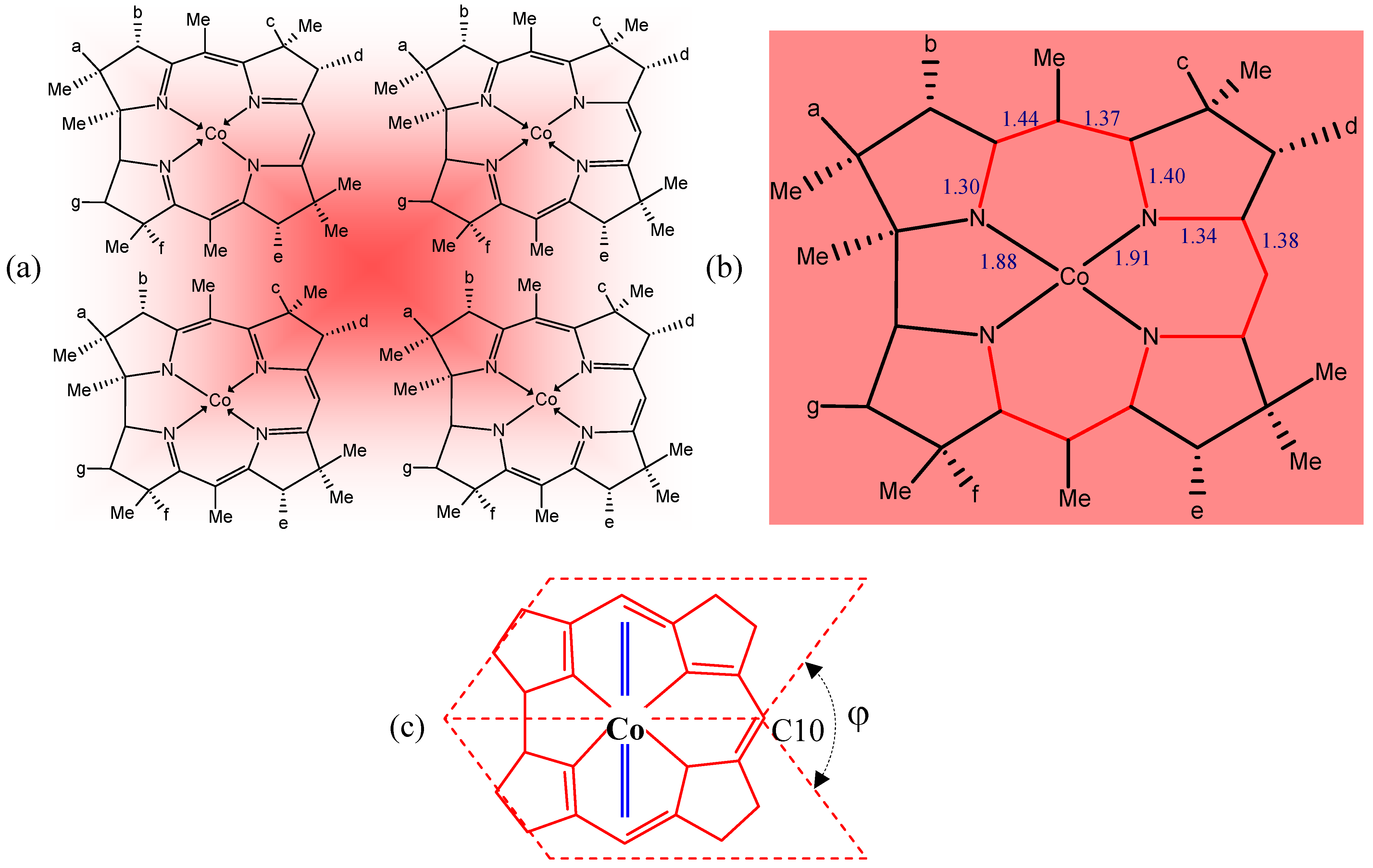
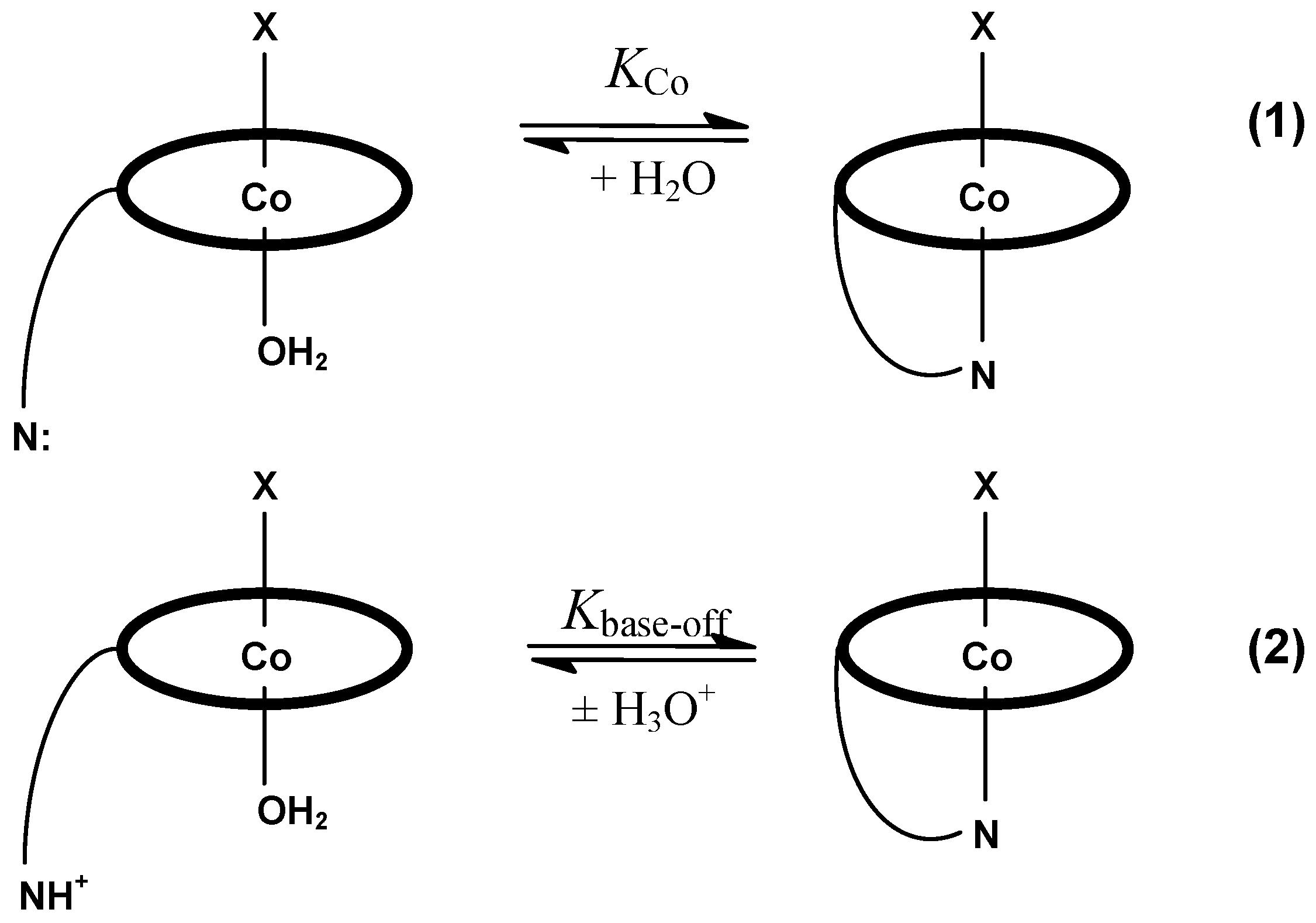
3. Relevant Structural Aspects of Cobalamins
4. Some Structure/Properties Relationships
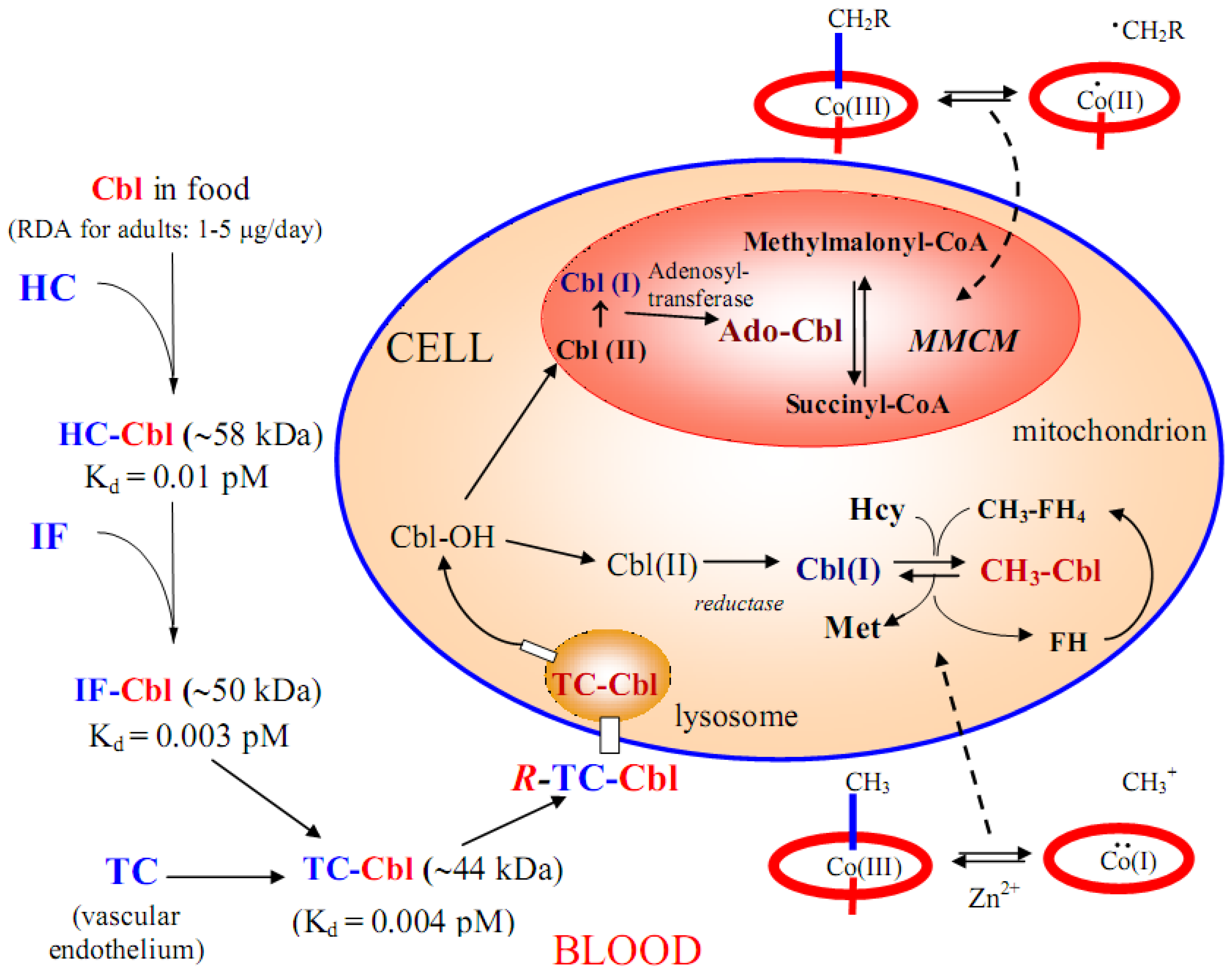
5. Absorption, Transport and Cellular Uptake of B12
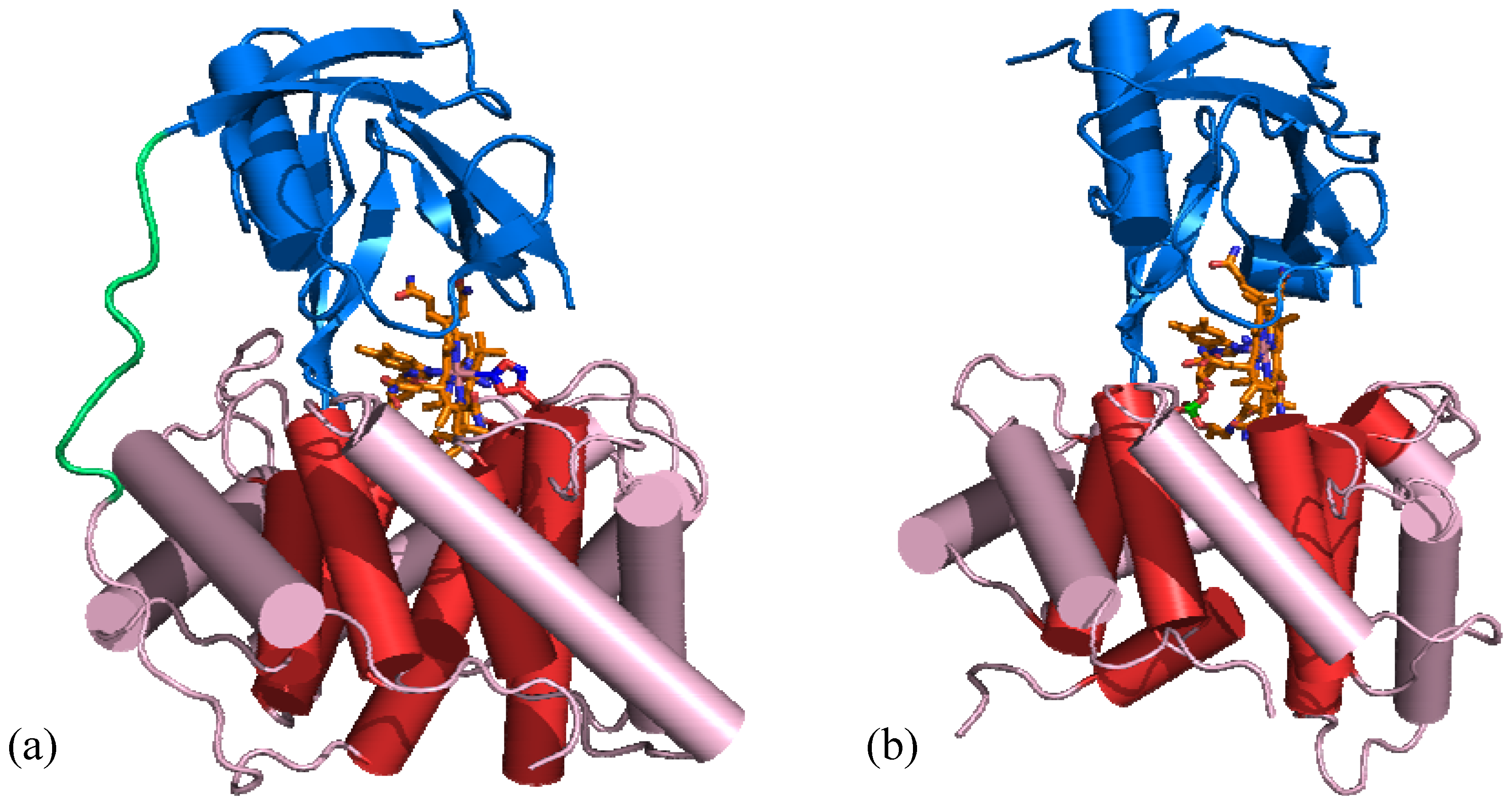
5.1. B12 transport proteins in mammals
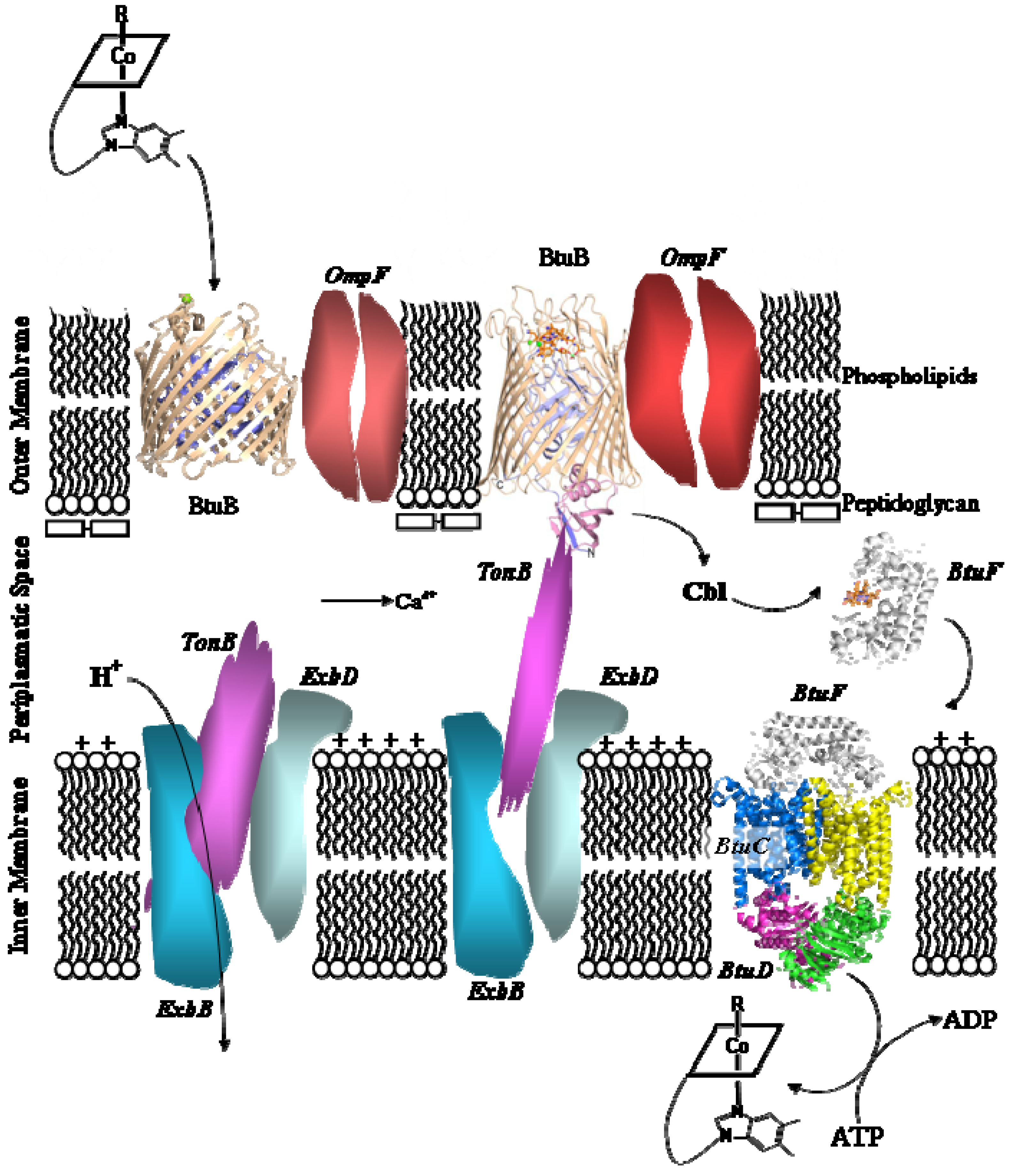
5.2. B12 transport proteins in bacteria
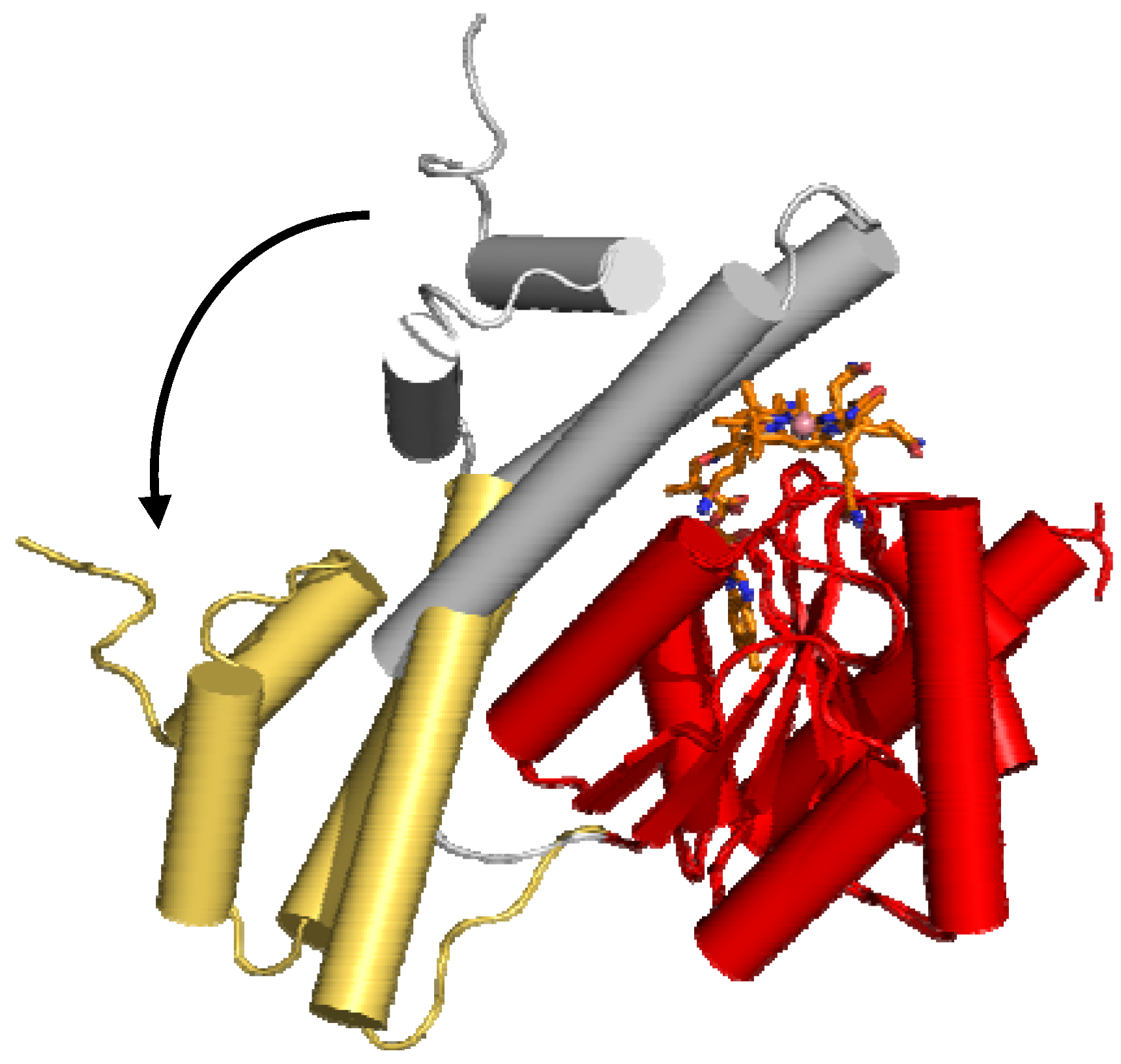
6. B12 Dependent Enzymes
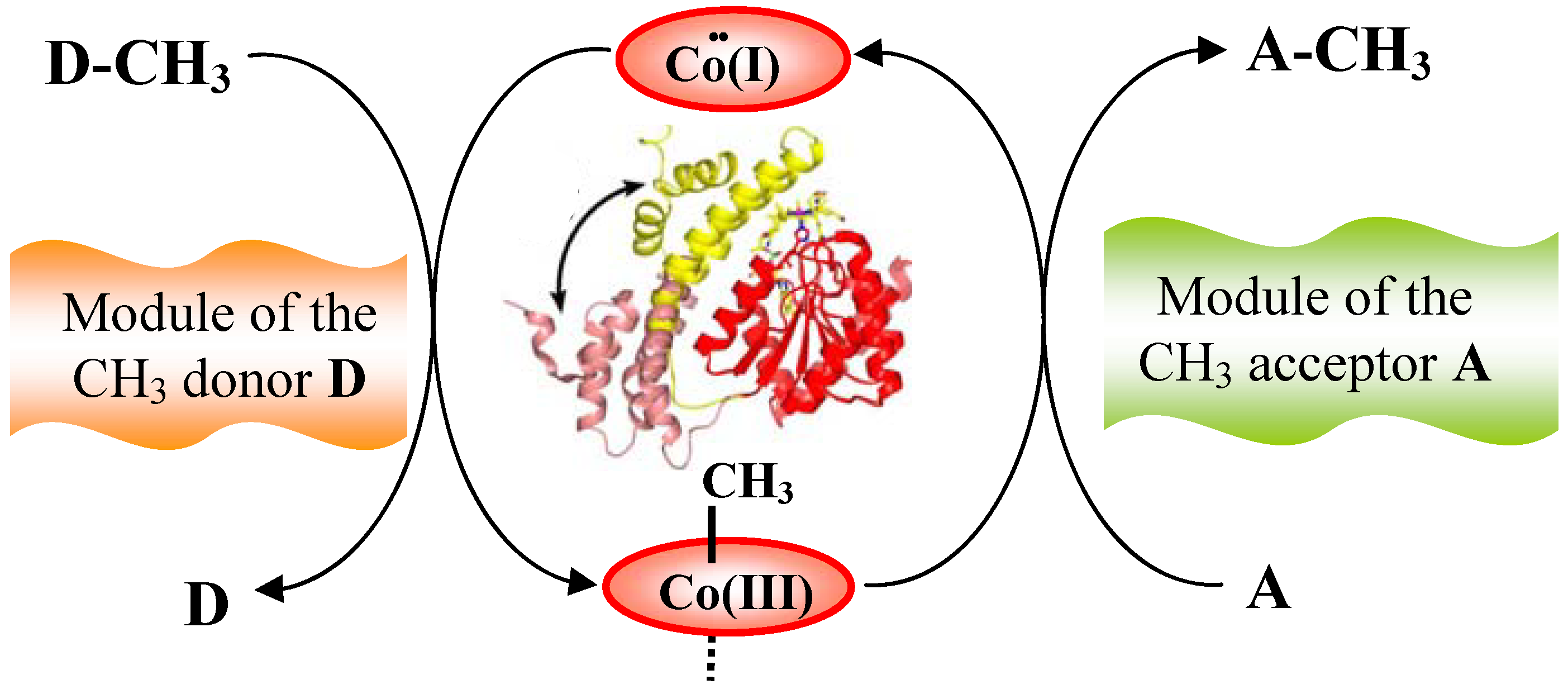
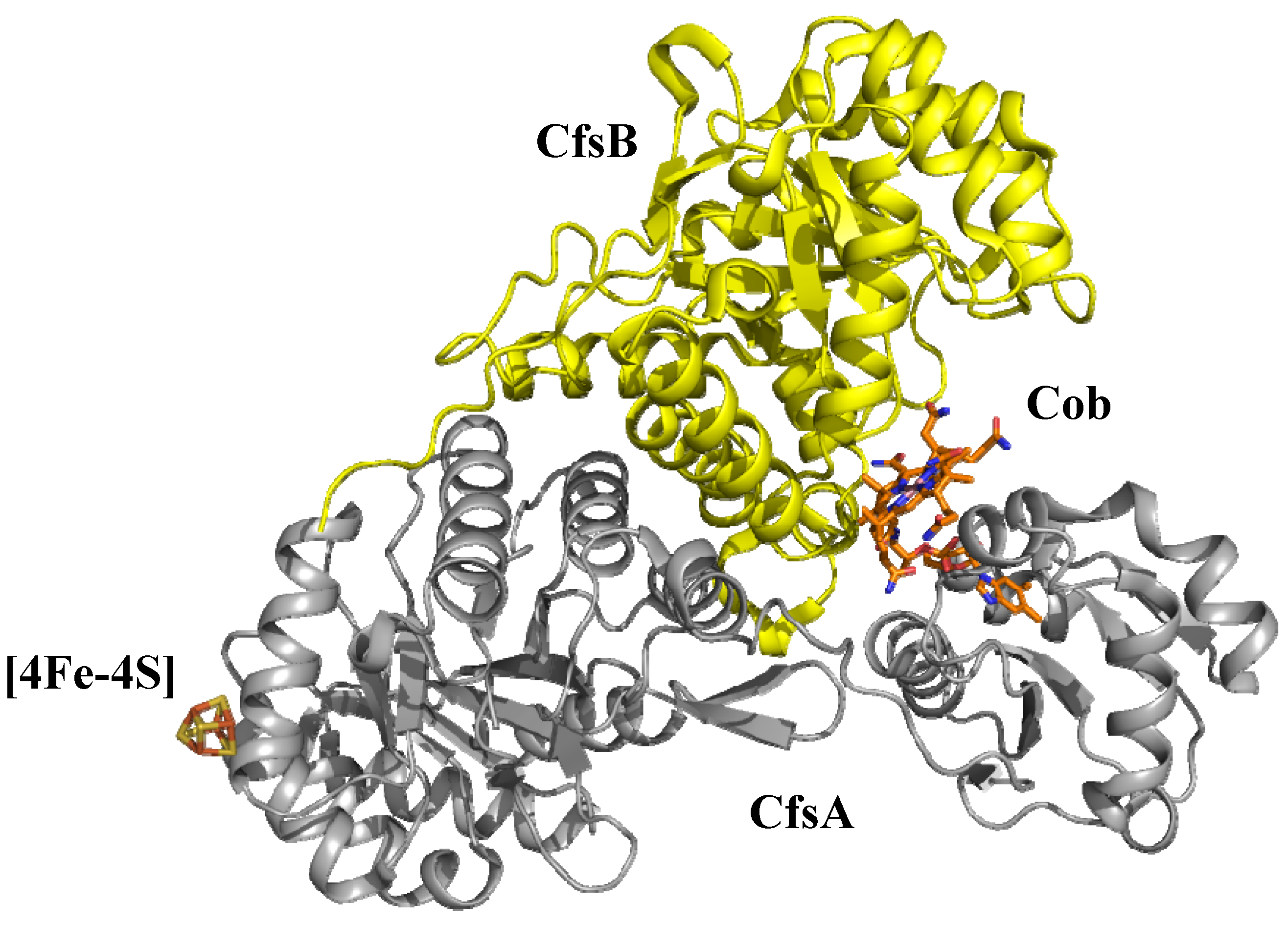
6.1. Corrinoid-dependent methyltransferase
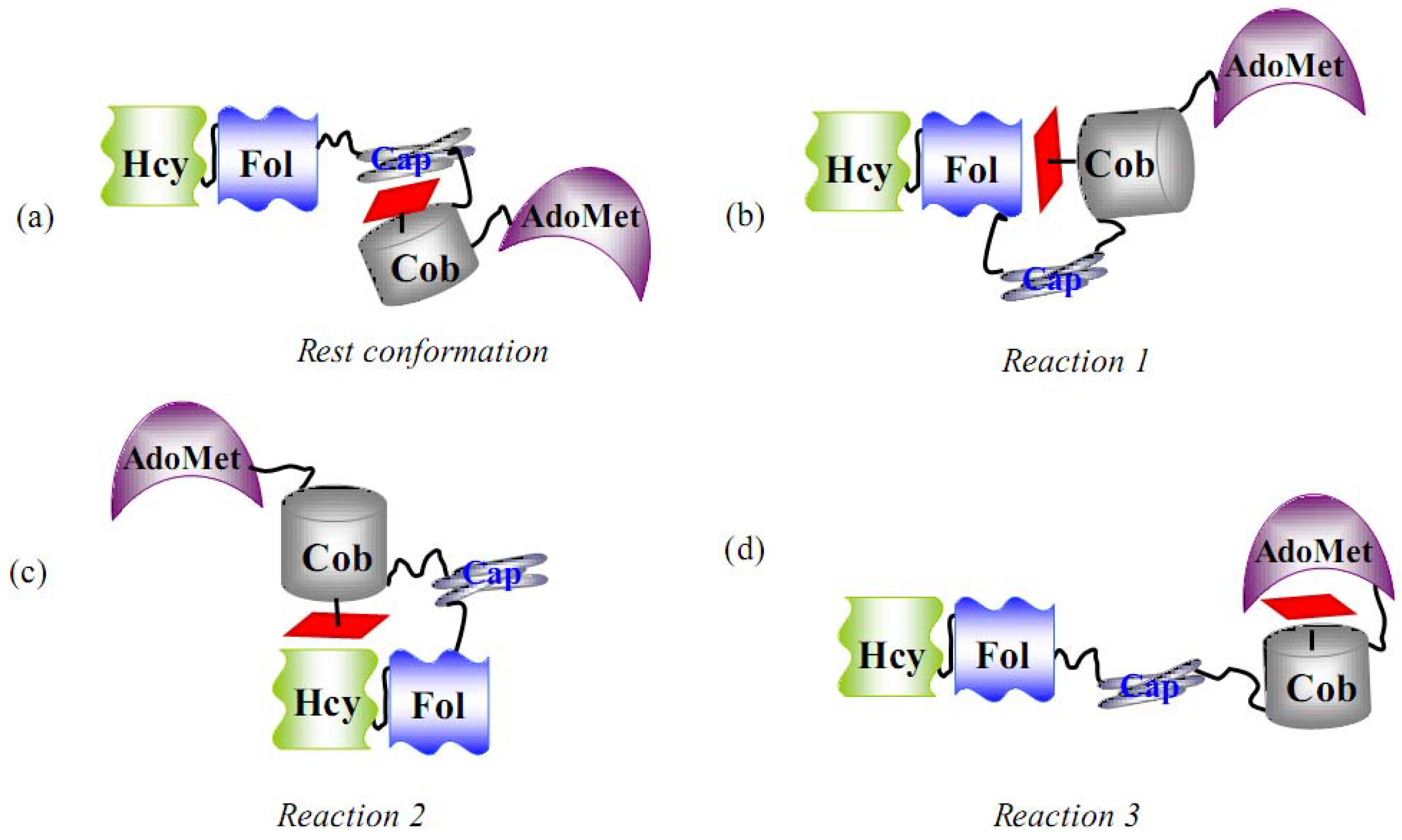
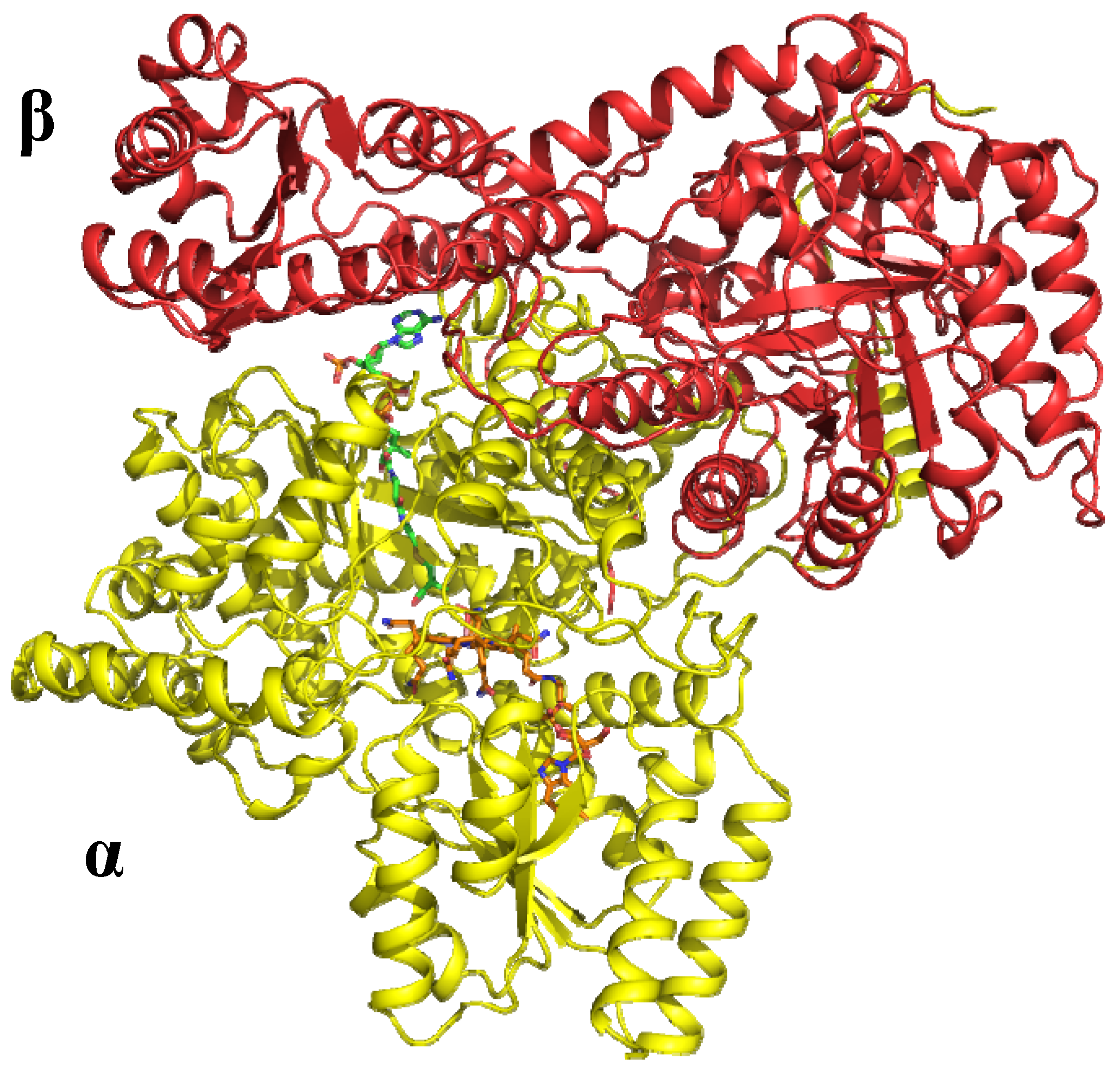
6.1.1. MeCbl methionine synthase (MetH)

6.1.2. Corrinoid-dependent methyltransferase in archaea and bacteria
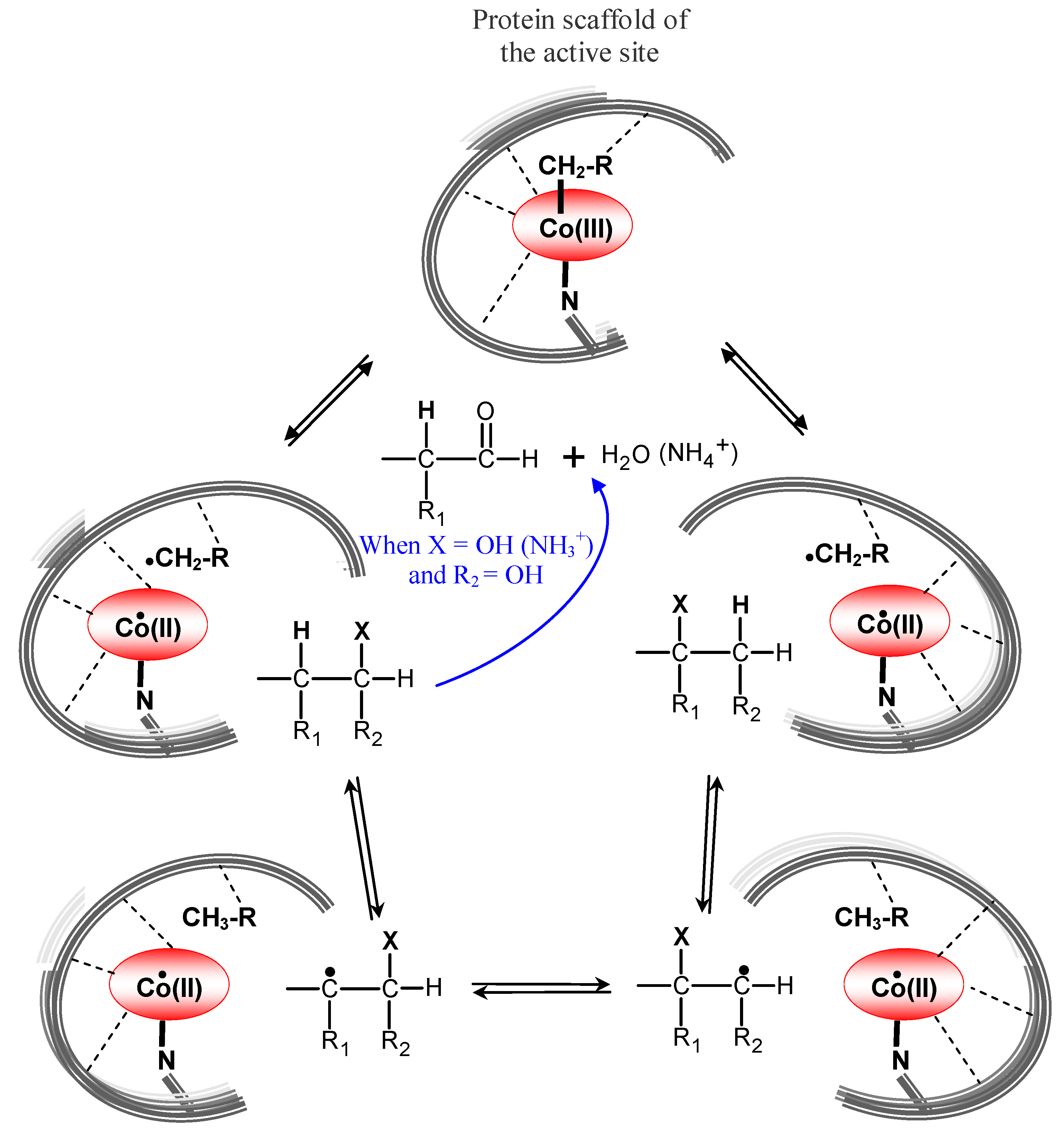
6.2. AdoCbl-dependent enzymes
6.3. Adenosyltransferase
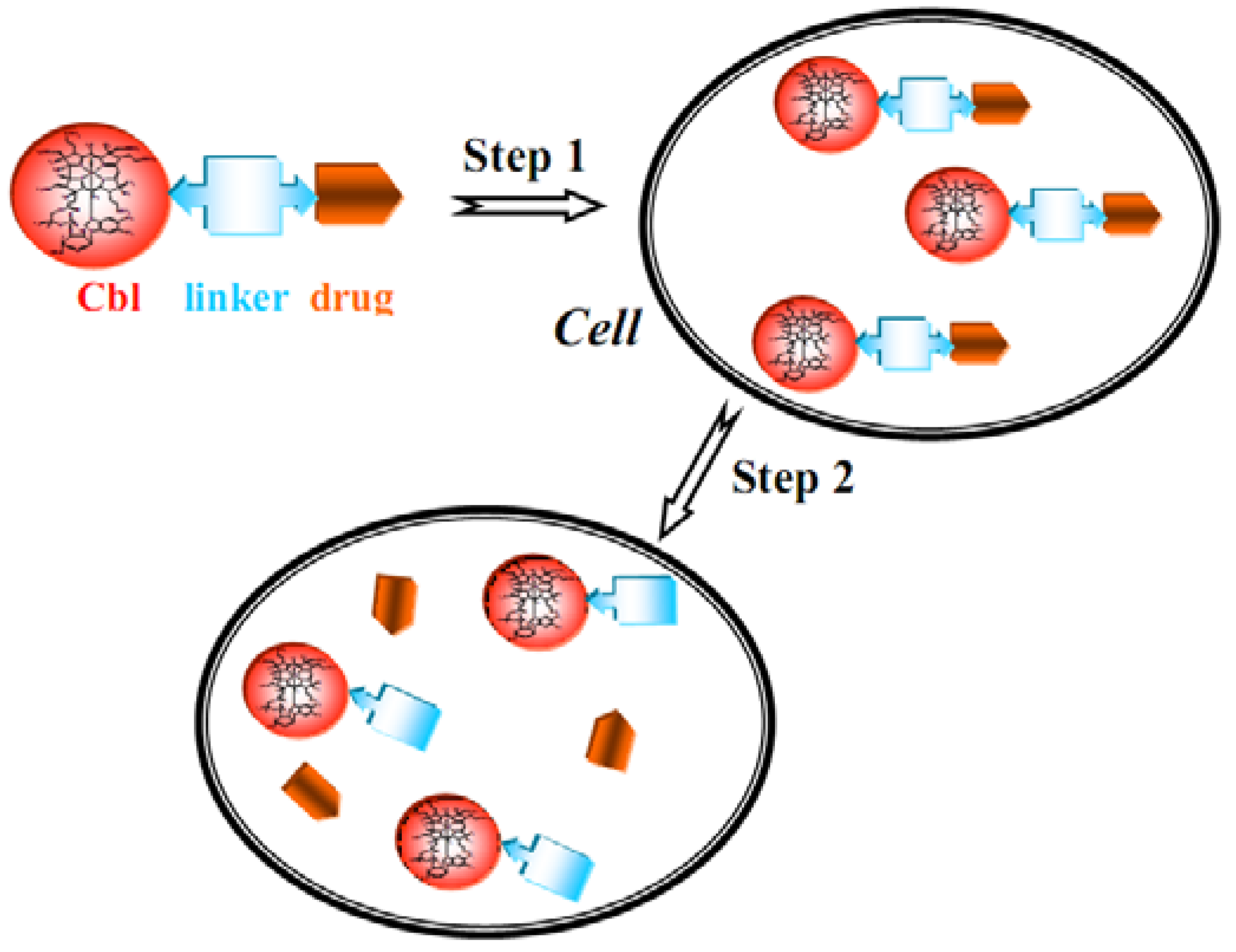
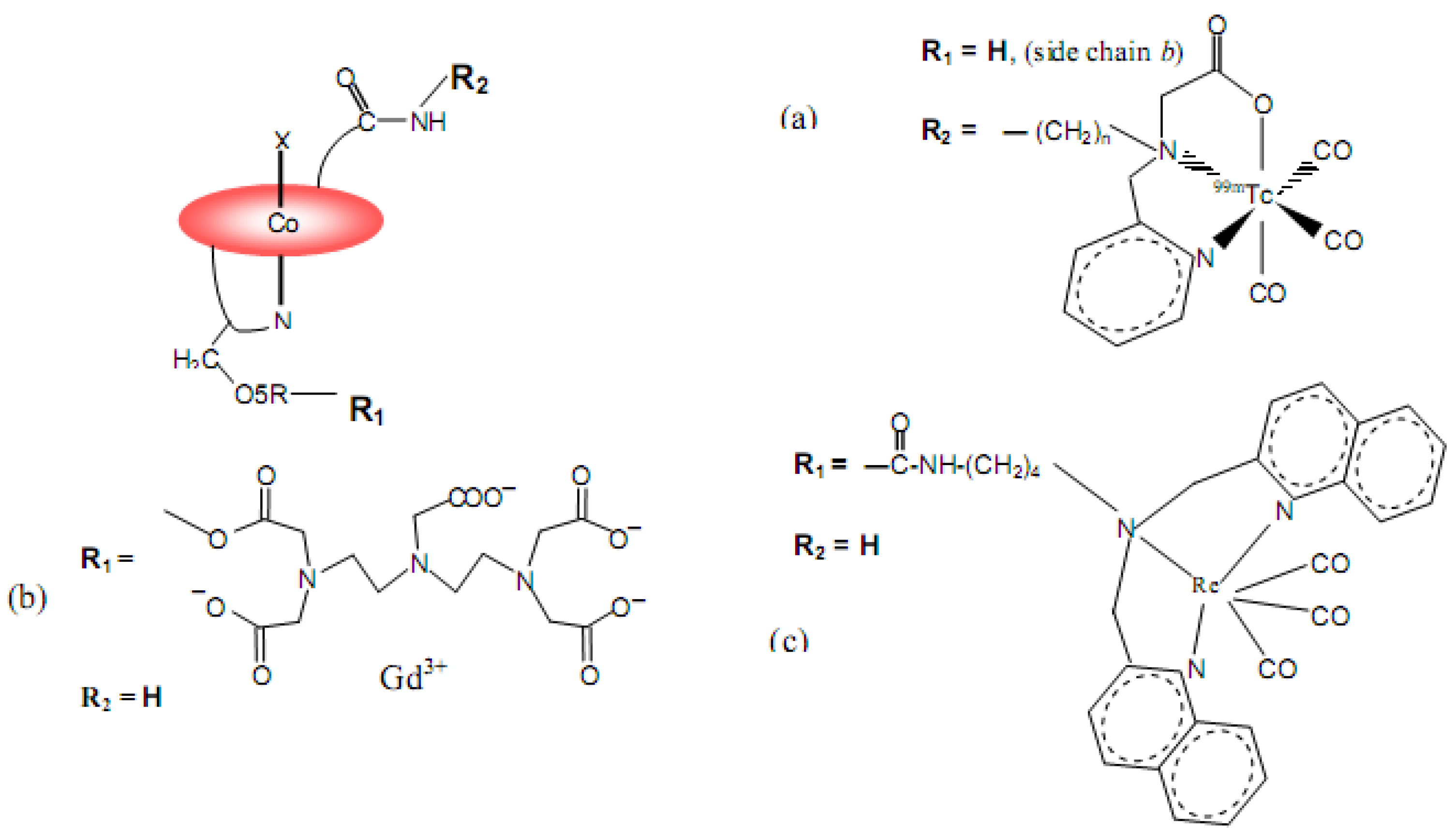
7. B12 Bioconjugates
8. Conclusions
Acknowledgments
References and Notes
- Folkers, K. History of Vitamin B12: Pernicious Anemia to Crystalline Cyanocobalamin. In Vitamin B12; Dolphin, D., Ed.; John Wiley & Sons: New York, NY, USA, 1982; Volume I, pp. 1–15. [Google Scholar]
- Hodgkin, D.C. New and Old Problem in the Structure Analysis of Vitamin B12. In Vitamin B12, Proceedings of the Third European Symposium on Vitamin B12 and Intrinsic Factors, Zurich, Switzerland, March 1999; Zagalak, B., Friedrich, W., Eds.; Walter de Gruyter: Berlin, Germany, 1979; pp. 19–36. [Google Scholar]
- Woodward, R.B. Synthetic vitamin B12. In Vitamin B12, Proceedings of the Third European Symposium on Vitamin B12 and Intrinsic Factors, Zurich, Switzerland, March 1999; Zagalak, B., Friedrich, W., Eds.; Walter de Gruyter: Berlin, Germany; 1979; pp. 37–88. [Google Scholar]
- Kräutler, B. Vitamin B12 and B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 3–43. [Google Scholar]
- Hogenkamp, H.P.C. Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 3–8. [Google Scholar]
- Brown, K.L. Chemistry and Enzymology of Vitamin B12. Chem. Rev. 2005, 105, 2075–2149. [Google Scholar] [CrossRef] [PubMed]
- Kräutler, B. Organometallic Chemistry of B12 Coenzymes. In Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Royal Society of Chemistry: Cambridge, UK, 2009; Volume 6, pp. 1–51. [Google Scholar]
- Matthews, R.G. Cobalamin- and Corrinoid-Dependent Enzymes. In Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Royal Society of Chemistry: Cambridge, UK, 2009; Volume 6, pp. 53–114. [Google Scholar]
- Battersby, A.R.; Leeper, F.J. Biosynthesis of B12 in the aerobic organism Pseudomonas denitrificans. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 507–535. [Google Scholar]
- Scott, A.I.; Roessner, C.A.; Santader, P.J. B12 Biosynthesis: the anaerobic pathway. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 537–556. [Google Scholar]
- Randaccio, L.; Geremia, S.; Wuerges, J. Crystallography of Vitamin B12 Proteins. J. Organometal. Chem. 2007, 692, 1198–1215. [Google Scholar] [CrossRef]
- Fedosov, S.N.; Fedosova, N.U.; Kräutler, B.; Nexø, E.; Petersen, T.E. Mechanisms of discrimination between cobalamins and their natural analogues during binding to the specific B12-transporting proteins. Biochemistry 2007, 46, 6446–6458, and references therein. [Google Scholar] [CrossRef] [PubMed]
- Wuerges, J.; Garau, G.; Geremia, S.; Fedosov, S.N.; Petersen, T.E.; Randaccio, L. Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc. Natl. Acad. Sci. USA 2006, 103, 4386–4391. [Google Scholar] [CrossRef] [PubMed]
- Mathews, F.S.; Gordon, M.M.; Chen, Z.; Rajashankar, K.R.; Ealick, S.E.; Alpers, D.H.; Sukumar, N. Crystal structure of human intrinsic factor: cobalamin complex at 2.6-Å resolution. Proc. Natl. Acad. Sci. USA 2007, 104, 17311–17316. [Google Scholar] [CrossRef] [PubMed]
- Russell-Jones, G.; McTavish, K.; McEwan, J.; Rice, J.; Novotnik, D. Vitamin-mediated targeting as a potential mechanism to increase drug uptake by tumours. J. Inorg. Biochem. 2004, 98, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Waibel, R.; Treichler, H.; Schaefer, N.G.; van Staveren, D.R.; Mudwiler, S.; Kunze, S.; Küenzi, M.; Alberto, R.; Nüesch, J.; Knuth, A.; Moch, H.; Schibli, R.; Shubiger, P.A. New Derivatives of Vitamin B12 Show Preferential Targeting of Tumours. Cancer Res. 2008, 68, 2904–2911. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.K.; Fairchild, T.J.; Doyle, R.P. Travelling the Vitamin B12 Pathway: Oral Delivery of Protein and Peptide Drugs. Angew. Chem. Int. Ed. 2009, 48, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. The acetogenic corrinoid proteins. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 633–653. [Google Scholar]
- Sauer, K.; Thauer, R.K. The role of corrinoids in methanogenesis. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 655–680. [Google Scholar]
- Koutmos, M.; Pejchal, R.; Bomer, T.M.; Matthews, R.G.; Smith, J.L.; Ludwig, M.L. Metal active site elasticity linked to activation of homocysteine in methionine synthase. Proc. Natl. Acad. Sci. USA 2008, 105, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M.; Mulliez, E. Ribonucleotide reductases. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 731–756. [Google Scholar]
- Wolhfarth, G.; Diekert, G. Reductive dehalogenases. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 871–893. [Google Scholar]
- Butler, P.; Ebert, M.O.; Lyskowski, A.; Gruber, K.; Kratky, C.; Kräutler, B. Vitamin B12: A Methyl Group without a Job? Angew. Chem. Int. Ed. 2006, 45, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Randaccio, L. Vitamin B12 Coenzyme Models: Perspectives on Recent Developments in the Chemistry of the Cobaloximes and Related Models. Comments Inorg. Chem. 1999, 21, 327–376, and references therein. [Google Scholar] [CrossRef]
- Randaccio, L.; Geremia, S.; Nardin, G.; Wuerges, J. X-ray structural chemistry of cobalamins. Coord. Chem. Rev. 2006, 250, 1332–1350. [Google Scholar] [CrossRef]
- Hogenkamp, H.P.C. Reactions of Alkyl Ligands Coordinated to Cobalamins and Cobaloximes. In Vitamin B12; Dolphin, D., Ed.; John Wiley & Sons: New York, NY, USA, 1982; Volume I, pp. 295–323. [Google Scholar]
- Luo, L.B.; Li, G.; Chen, H.L.; Fu, S.W.; Zhang, S.Y. Laser-induced photoacoustic calorimetric determination of enthalpy and volume changes in photolysis of 5’-deoxyadenosylcobalamin and methylcobalamin. J. Chem. Soc. Dalton Trans. 1998, 2103–2107. [Google Scholar] [CrossRef]
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Structural aspects of B12 chemistry and biochemistry: From simple models to proteins. Trends Inorg. Chem. 2009, 11, 1–19. [Google Scholar]
- Hannibal, L.; Smith, C.A.; Chavez, R.A.; Jacobsen, D.W.; Brasch, N. E Nitroxylcob(III)alamin: Synthesis and X-ray Structural Characterization. Angew. Chem. Int. Ed. 2007, 46, 5140–5143. [Google Scholar] [CrossRef] [PubMed]
- Brasch, N.E.; Kent State University, Kent, USA. Personal communication, 2009.
- Hannibal, L.; Smith, C.A.; Smith, J.A.; Axhemi, A.; Miller, A.; Wang, S.; Brasch, N.E.; Jacobsen, D.W. High Resolution Crystal Structure of the Methylcobalamin Analogues Ethylcobalamin and Butylcobalamin by X-ray Synchrotron Diffraction. Inorg. Chem. 2009, 48, 6615–6622. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, H.A.; Hannibal, L.; Jacobsen, D.W.; Brown, K.L.; Marques, M.M.; Brasch, N.E. NMR spectroscopy and molecular modelling studies of nitrosylcobalamin: further evidence that the deprotonated, base-off form is important for nitrosylcobalamin in solution. Dalton Trans. 2009, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Kuta, J.; Wuerges, J.; Randaccio, L.; Kozlowski, P.M. Axial Bonding in Alkylcobalamins: DFT Analysis of the Inverse versus Normal Trans Influence. J. Phys. Chem. A 2009, 113, 11604–11612. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Peck-Siler, S. Heteronuclear NMR Studies of Cobalamins. 9. Temperature-Dependent NMR of Organocobalt Corrin Enriched in 13C in the Organic Ligand and the Thermodynamics of Base-On/Base-Off Reactions. Inorg. Chem. 1988, 27, 3548–3555. [Google Scholar] [CrossRef]
- Pratt, J.M. The Roles of Co, Corrins, and Protein. I. Co-Ligand Bonding and the Trans Effect. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 73–112. [Google Scholar]
- Alpers, D.H.; Russel-Jones, G.J. Intrinsic factor, haptocorrin and their receptors. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 411–440. [Google Scholar]
- Rothenberg, S.P.; Quadros, E.V.; Regec, A. Transcobalamin II. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 441–473. [Google Scholar]
- Andersen, C.B.; Madsen, M.; Storm, T.; Moestrup, S.K.; Andersen, G.R. Structural basis for receptor recognition of vitamin-B12-intrinsic factor complexes. Nature 2010, 464, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Nexø, E. Cobalamin Binding Proteins. In Vitamin B12 and B12 Proteins; Kräutler, B., Arigoni, D., Golding, B.T., Eds.; Wiley-VCH: Weinheim, Germany, 1998; pp. 461–471. [Google Scholar]
- Quadros, E.V.; Nakayama, Y.; Sequeira, J.M. The protein and the gene encoding the receptor for the cellular uptake of transcobalamin-bound cobalamin. Blood 2009, 113, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R. B12 Trafficking in Mammals: A Case for Coenzyme Escort Service. ACS Chem. Bio. 2006, 1, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Gherasim, C.; Padovani, D. The tinker, tailor, soldier in intracellular B12 trafficking. Curr. Opin. Chem. Biol. 2009, 13, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Bradbeer, C. Cobalamin Transport in Bacteria. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 489–506. [Google Scholar]
- Fedosov, S.N.; Berglund, L.; Fedosova, N.U.; Nexø, E.; Petersen, T.E. Comparative analysis of cobalamin binding kinetics and ligand protection for intrinsic factor, transcobalamin, and haptocorrin. J. Biol. Chem. 2002, 277, 9989–9996. [Google Scholar] [CrossRef] [PubMed]
- Wuerges, J.; Geremia, S.; Randaccio, L. Structural study on ligand specificity of human vitamin B12 transporters. Biochem. J. 2007, 403, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Fedosov, S.N.; Fedosova, N.U.; Berglund, L.; Moestrup, S.K.; Nexø, E.; Petersen, T.E. Composite Organization of the Cobalamin Binding and Cubilin Recognition Sites of Intrinsic Factor. Biochemistry 2005, 44, 3604–3614. [Google Scholar] [CrossRef] [PubMed]
- Wuerges, J.; Geremia, S.; Fedosov, S.N.; Randaccio, L. Vitamin B12 Transport Proteins: Crystallographic Analysis of β-Axial Ligand Substitution in Cobalamin Bound to Transcobalamin. IUBMB Life 2007, 59, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Hogenkamp, H.P.; Collins, D.A.; Grissom, C.B.; West, F.G. Diagnostic and Therapeutic Analogues of Cobalamins. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 385–410. [Google Scholar]
- Chimento, D.P.; Mohanty, A.K.; Kadner, R.J.; Wiener, M.C. Substrate-induced transmembrane signalling in the cobalamin transporter BtuB. Nature Struct. Biol. 2003, 10, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Shultis, D.D.; Purdy, M.D.; Banchs, C.N.; Wiener, M.C. Outer Membrane Active Transport: Structure of the BtuB:TonB Complex. Science 2006, 312, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Locher, K.P.; Lee, A.T.; Rees, D.C. The E. coli BtuCD Structure: a Framework for ABC Transporter Architecture and Mechanism. Science 2002, 296, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Karpowich, N.K.; Huang, H.H.; Smith, P.C.; Hunt, J.F. Crystal Structures of the BtuF Periplasmatic-binding Protein for Vitamin B12 Suggest a Functionally Important Reduction in Protein Mobility upon Ligand Binding. J. Biol. Chem. 2003, 278, 8429–8434. [Google Scholar] [CrossRef] [PubMed]
- Borths, E.L.; Locher, K.P.; Lee, A.T.; Rees, D.C. The structure of Escherichia coli BtuF and binding its cognate ATP binding cassette transporter. Proc. Natl. Acad. Sci. USA 2002, 99, 16642–16647. [Google Scholar] [CrossRef] [PubMed]
- Hvorup, R.N.; Goetz, B.A.; Niederer, M.; Hollenstein, K.; Perozo, E.; Locher, K.P. Asymmetry in the Structure of the ABC Transporter-Binding Protein Complex BtuCD-BtuF. Science 2007, 317, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
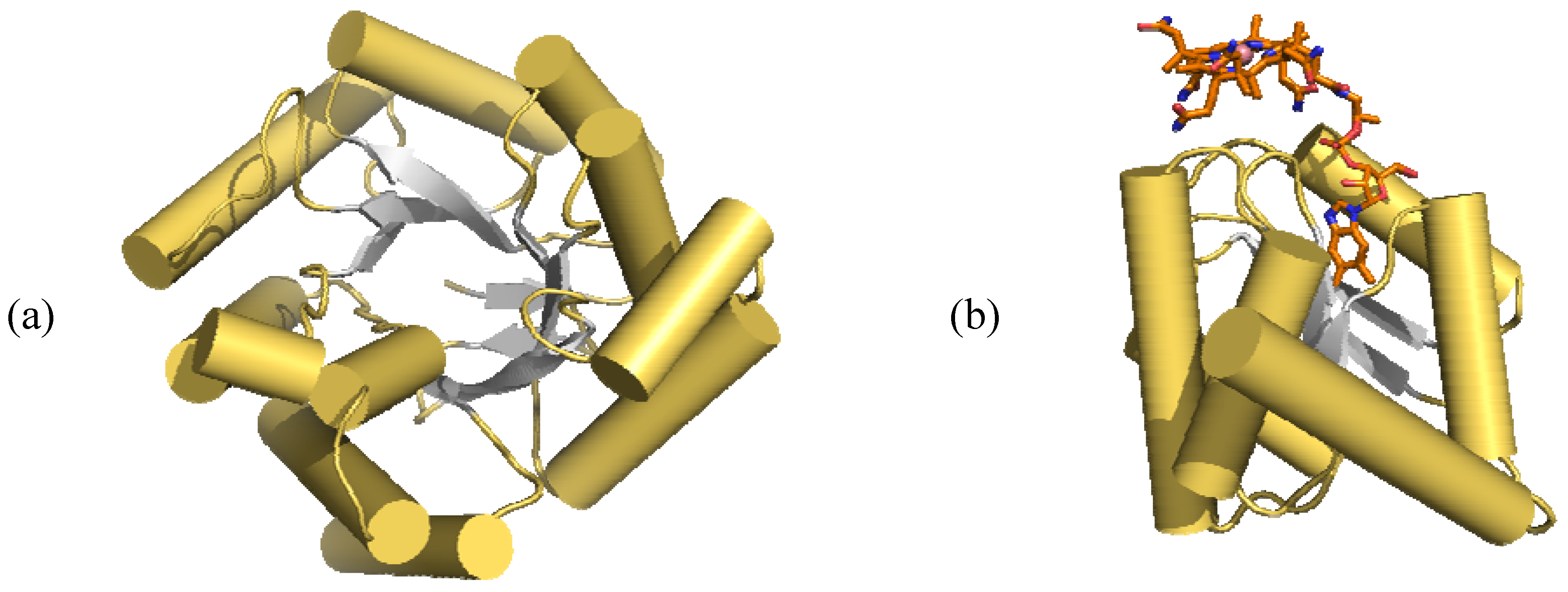
- Drennan, C.L.; Huang, S.; Drummond, J.T.; Mattews, R.G.; Ludwig, M.L. How a protein binds B12: A 3.0 X-ray structure of B12-binding domain of methionine synthase. Science 1994, 266, 1699–1704. [Google Scholar] [CrossRef]
- Mancia, F.; Keep, N.H.; Nakagawa, A.; Leadlay, P.F.; McSweeney, S.; Rasmussen, B.; Bösecke, P.; Diat, O.; Evans, P.R. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. [Google Scholar] [CrossRef]
- Evans, J.C.; Huddler, D.P.; Hilgers, M.T.; Romanchuk, G.; Matthew, R.G.; Ludwig, M.L. Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc. Natl. Acad. Sci. USA 2004, 101, 3729–3736. [Google Scholar] [CrossRef] [PubMed]
- Koutmos, M.; Pejchal, R.; Bomer, T.M.; Matthew, R.G.; Smith, J.L.; Ludwig, M.L. Metal active site elasticity linked to activation of homocystine in methionine synthase. Proc. Natl. Acad. Sci. USA 2008, 105, 3286–3291. [Google Scholar] [CrossRef] [PubMed]
- March, E.N.G.; Holloway, D.E. Cloning and sequencing of glutamate mutase component S from Clostridium tetanomorphum. FEBS Lett. 1992, 310, 167–170. [Google Scholar]
- Bandarian, V.; Pattridge, K.A.; Lennon, B.W.; Huddler, D.P.; Matthews, R.G.; Ludwig, M.L. Domain alternation switches B12-dependent methionine synthase to the activation conformation. Nat. Struct. Biol. 2002, 9, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Koutmos, M.; Pattridge, K.A.; Ludwig, M.L.; Matthew, R.G. A disulfide-stabilized conformer of methionine synthase reveals an unexpected role for the histidine ligand of the cobalamin cofactor. Proc. Natl. Acad. Sci. USA 2008, 105, 4115–4120. [Google Scholar] [CrossRef] [PubMed]
- Bandarian, V.; Matthews, R.G.; Ludwig, M.L. Factors modulating conformational equilibria in large modular proteins: A case study with cobalamin-dependent methionine synthase. Proc. Natl. Acad. Sci. USA 2003, 100, 8156–8163. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, A.S.; Matthews, R.G. Ligand Trans Influence Governs Conformation in Cobalamin-Dependent Methionine Synthase. Biochemistry 2007, 46, 12382–12392. [Google Scholar] [CrossRef] [PubMed]
- Hagemeier, C.H.; Krüer, M.; Thauer, R.K.; Warkentin, B.; Ermler, U. Insight into the mechanism of biological methanol activation based on the crystal structure of the methanol-cobalamin methyltransferase complex. Proc. Natl. Acad. Sci. USA 2006, 103, 18917–18922. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Thauer, R.K. The Role of Corrinoids in Methanogenesis. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 655–679. [Google Scholar]
- Svetlitchnaia, T.; Svetlitchnyi, V.; Meyer, O.; Dobbek, H. Structural insights into methytransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14331–14336. [Google Scholar] [CrossRef] [PubMed]
- Randaccio, L.; Bresciani-Pahor, N.; Zangrando, E.; Marzilli, L.G. Structural Properties of Organocobalt Coenzyme B12 Models. Chem. Soc. Rev. 1989, 18, 225–250. [Google Scholar] [CrossRef]
- Toraya, T. Radical catalysis of B12 enzymes: structure, mechanism, inactivation, and reactivation of diol and glycerol dehydratases. Cell. Mol. Life Sci. 2000, 57, 106–127. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R. Radical Skeleton Rearrangements: Catalysis by Coenzyme B12-Dependent Mutases. Chem. Rev. 2003, 103, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Toraya, T. Radical Catalysis in Coenzyme B12-Dependent Isomerization (eliminating) Reactions. Chem. Rev. 2003, 103, 2095–2127. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A.; Essenberg, M.K.; Abeles, R.H. Studies on the Mechanism of Hydrogen Transfer in the Cobamide Coenzyme-dependent Dioldehydratase Reaction. J. Biol. Chem. 1967, 242, 5369–5377. [Google Scholar] [PubMed]
- Retey, J.; Arigoni, D. Coenzym B12 als gemeinsamer Wassestoffüberträger der Dioldehydratase- und der Methylmalonyl-CoA-mutase-Reaktion. Experientia 1966, 22, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Sintchak, M.D.; Arjara, G.; Kellog, B.A.; Stubbe, J.; Drennan, C.L. The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nature Struct. Biol. 2002, 9, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Buckel, W.; Golding, B.T.; Kratky, C. Stabilisation of Methylene Radicals by Cob(II)alamin in Coenzyme B12 dependent Mutases. Chem. Eur. J. 2006, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, P.M.; Kamachi, T.; Toraya, T.; Yoshizawa, K. Does Cob(II)alamin Act as a Conductor in Coenzyme B12 Dependent Mutases. Angew. Chem. Int. Ed. 2007, 46, 980–983. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Kozlowski, P.M. Role of the Tyrosine Residue in the Activation of the Co-C Bond in Coenzyme B12-Dependent Enzymes: Another Case of Proton-Coupled Electron Transfer. J. Phys.Chem. B 2009, 113, 9050–9054. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, D.S.; Fenton, W.A. Methylmalonyl-CoA mutase. In Chemistry and Biochemistry of B12; Banerjee, R., Ed.; John Wiley & Sons: New York, NY, USA, 1999; pp. 367–384. [Google Scholar]
- Stich, T.A.; Buan, N.R.; Escalante-Semerena, J.C.; Brunold, T.C. Spectroscopic and Computational Studies of the ATP:Corrinoid Adenosyltransferase (CobA) from Salmonella enterica: Insights into the Mechanism of Adenosylcobalamin Biosynthesis. J. Am. Chem. Soc. 2005, 127, 8710–8719. [Google Scholar] [CrossRef] [PubMed]
- Schubert, H.L.; Hill, C.P. Structure of ATP-bound human ATP: Cobalamin Adenosyltransferase. Biochemistry 2006, 45, 15188–15196. [Google Scholar] [CrossRef] [PubMed]
- St. Maurice, M.; Mera, P.E.; Taranto, M.P.; Sesma, F.; Escalante-Semerena, J.; Rayment, I. Structural characterization of the active site of the PduO-type ATP:Co(I)rrinoid Adenosyltransferase from Lactobacillus reuteri. J. Biol. Chem. 2007, 282, 2596–2605. [Google Scholar] [CrossRef] [PubMed]
- St. Maurice, M.; Mera, P.; Park, K.; Brunold, T.C.; Escalante-Semerena, J.; Rayment, I. Structural Characterization of a Human-Type Corrinoid Adenosyltransferase Confirms that Coenzyme B12 is synthesized trough a Four-Coordinate Intermediate. Biochemistry 2008, 47, 5755–5766. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.E.; St. Maurice, M.; Rayment, I.; Escalante-Semerena, J. Residue Phe112 of the Human-Type Corrinoid Adenosyltransferase (PduO) Enzyme of Lactobacillus reuteri Is Critical to the formation of the Four-Coordinate Co(II) Corrinoid Substrate and to the Activity of the Enzyme. Biochemistry 2009, 48, 3138–3145. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.K.; Fairchild, T.J.; Doyle, R.P. Traveling the Vitamin B12 Pathway: Oral Delivery of Protein and Peptide Drugs. Angew. Chem. Int. Ed. 2009, 48, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.K.; Allis, D.G.; Smith, P.R.; Fairchild, T.J.; Doyle, R.P. Exploring the Implications of Vitamin B12 Conjugation to Insulin on Insulin Receptor Binding. ChemMedChem 2009, 4, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.; Kohli, D.V.; Jain, S.D. Vitamin B12-mediated transport: a potential tool for tumor targeting of antineoplastic drugs and imaging agents. Crit. Rev. Ter. Drug Carrier Syst. 2008, 25, 347–379. [Google Scholar] [CrossRef]
- Siega, P.; Wuerges, J.; Arena, F.; Gianolio, E.; Fedosov, S.N.; Dreos, R.; Geremia, S.; Aime, S.; Randaccio, L. Release of Toxic Gd3+ Ions to Tumour Cells by Vitamin B12 Bioconjugates. Chem. Eur. J. 2009, 15, 7980–7989. [Google Scholar] [CrossRef] [PubMed]
- Viola-Villegas, N.; Rabideau, A.E.; Bartholoma, M.; Zubieta, J.; Doyle, R.P. Targeting the Cubilin Receptor through the Vitamin B12 Uptake Pathway: Cytotoxicity and Mechanistic Insight through Fluorescent Re(I) Delivery. J. Med. Chem. 2009, 52, 5253–5261. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not applicable. |

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dCo-Ca | dCo-NB3 a | Co-C-Cb | νCo-Cc | BDEd | |
| AdoCbl | 2.033(4) | 2.237(3) | 123.4(2) | 430 | 125 ± 8 |
| MeCbl | 1.979(4) | 2.162(4) | - | 506 | 155 ± 13 |
| X | pKbase-offa | KCo | ΔG0Co (kJ/mol) | Co-NB3b |
|---|---|---|---|---|
| NO | 5.1 | 1.9 | -1.59 | 2.349 (2)c |
| CH3CH2 | 4.16 | 2.00·10 | -7.42 | 2.232 (1)d |
| CH3CH2CH2 | 4.10 | 2.37·10 | -7.84 | / |
| Ado | 3.67 | 7.25·10 | -10.6 | 2.237 (3) |
| CH3 | 2.90 | 4.52·102 | -15.1 | 2.162 (4) |
| AdoPr | 3.31 | 1.77·102 | -12.8 | 2.212 (8) |
| CF3CH2 | 2.60 | 9.23·102 | -16.9 | / |
| CH2=CH | 2.4 | 1.5·103 | -18 | 2.165 (6) |
| cis ClCH=CH | 2.3 | 1.8·103 | -19 | 2.144 (5) |
| CF2H | 2.15 | 2.60·103 | -19.5 | 2.187 (7) |
| NCCH2 | 1.81 | 5.62·103 | -21.4 | / |
| CF3 | 1.44 | 1.32·104 | -23.5 | 2.05 (1) |
| CN | 0.10 | 2.88·105 | -31.1 | 2.041 (3) |
| H2O | -2.13 | 4.90·107 | -43.9 | 1.925 (2) |
| X | R | Enzyme | |
|---|---|---|---|
| Class I | |||
 | -CH(NH3+)CO2− | CO2− | GLM |
| -C(CO2−)=CH2 | CO2− | MGM | |
| COSCoA | CO2− | MMCM | |
| COSCoA | H | IBCM | |
| Class II | R | Enzyme | |
 | H, CH3, CH2OH | DD GD | |
 | H, CH3 | EAL | |
| Class III | R | Enzyme | |
 | −O2C(NH3+)HC(CH2)2- | LAM | |
| −O2C(NH3+)HCCH2- | OAM | ||
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins. Molecules 2010, 15, 3228-3259. https://doi.org/10.3390/molecules15053228
Randaccio L, Geremia S, Demitri N, Wuerges J. Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins. Molecules. 2010; 15(5):3228-3259. https://doi.org/10.3390/molecules15053228
Chicago/Turabian StyleRandaccio, Lucio, Silvano Geremia, Nicola Demitri, and Jochen Wuerges. 2010. "Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins" Molecules 15, no. 5: 3228-3259. https://doi.org/10.3390/molecules15053228
APA StyleRandaccio, L., Geremia, S., Demitri, N., & Wuerges, J. (2010). Vitamin B12: Unique Metalorganic Compounds and the Most Complex Vitamins. Molecules, 15(5), 3228-3259. https://doi.org/10.3390/molecules15053228