3. Experimental
3.1. General
Melting points were determined on an Electrothermal IA9000 series digital capillary melting point apparatus. IR spectra were run (KBr discs) on a Shimadzu FT spectrophotometer 1000.
1H- and
13C- NMR spectra were recorded in DMSO-d
6 (or in CDCl
3) on a JEOL ECP 400 NMR spectrometer operating at 400/100 MHz, with TMS as internal standard. DEPT and HETCOR experiments were recorded on a 500 MHz instrument (Bruker, J.F.B. 288) at King Saud University (Pharmacy Research Centre). Chemical shifts are given in δ (ppm) and coupling constants (
J) are given in Hz. The assignments of all carbons are made by comparison to
13C-NMR spectra of structurally related compounds [
12,
13] and theoretical grounds [
20,
21], and with the aid various modern NMR techniques in many cases. Electron impact (EI) MS spectra were recorded on a Shimadzu GCMSQP5050A spectrometer (DB-1 glass column 30 m × 0.25 mm, ionization energy 70 eV), at the Chemistry Department, College of Science, King Saud University. Antimicrobial and anti-cancer tests of some of the synthesized compounds were run in King Abdul Aziz Hospital for the National Guard, Al-hasa, Saudi Arabia and the Pharmacology Unit, National Cancer Institute, Cairo University, Egypt, respectively. The reactions were monitored by TLC, and the purity of the compounds were routinely checked by TLC silica gel plates while the spots were visualised by UV (Uvitec). The starting materials, ethyl 2-amino-4,5-disubstituted thiophene-3-carboxylates
1a-d and the 2-amino-4,5-disubstituted thiophene-3-carbonitrile
32, were prepared by condensation of ketones, elemental sulfur, ethyl cyanoacetate or malononitrile as described [
14,
15].
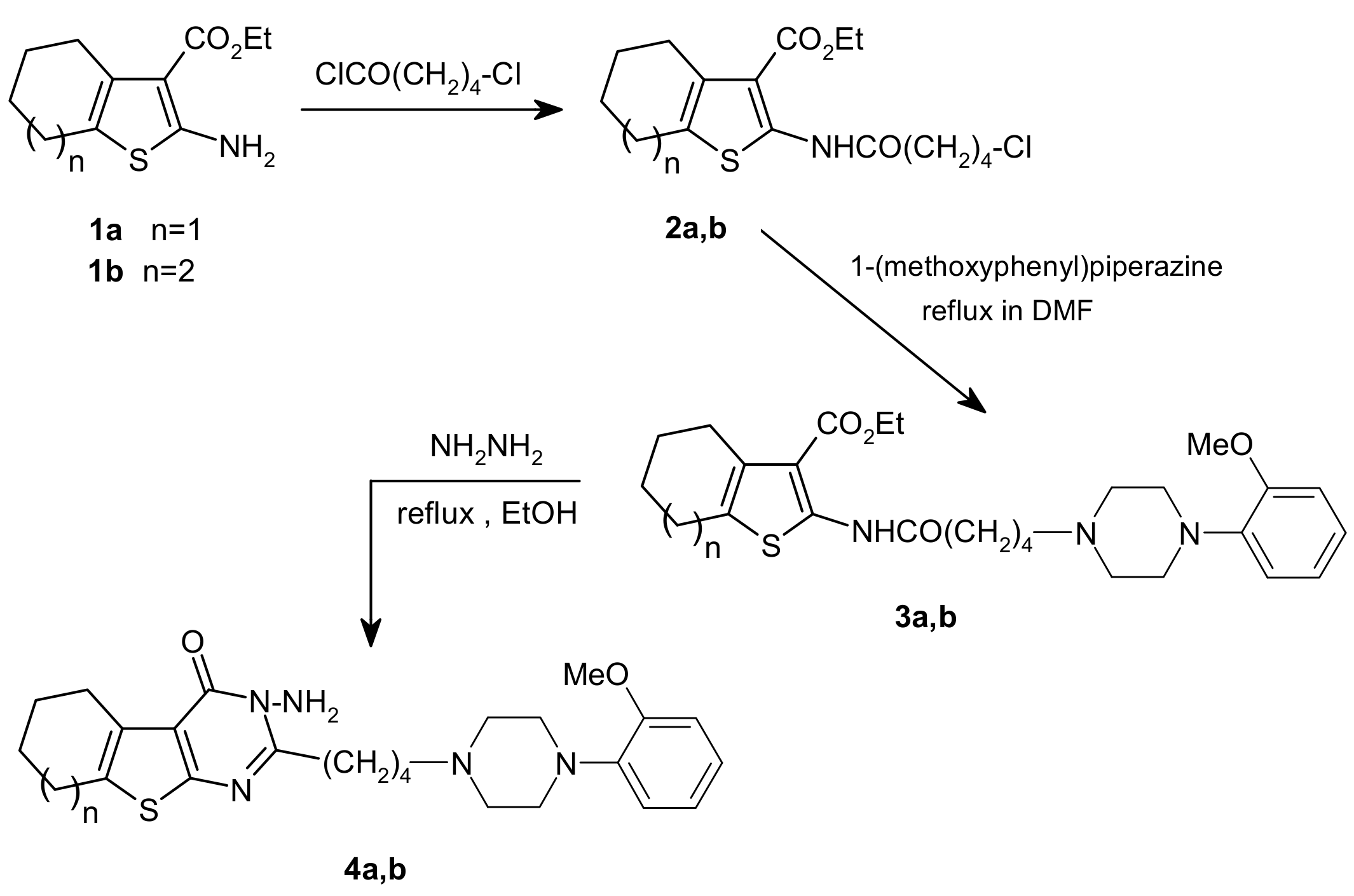
3.2. General procedure for synthesis of 2a,b
Compounds
2a,b were both prepared according to a method reported in the literature [
22] for similar compounds. 5-Chlorovaleryl chloride (1.3 mL, 10 mmol) was added to a solution of amino ester
1a,b (10 mmol) in chloroform (20 mL) and the solution was refluxed for 4 h. After cooling, the solution was concentrated under reduced pressure to give a dark oil. Addition of a small amount of water and ethanol yielded a solid that was collected, dried and recrystallized from ethanol.
2-(5-Chloropentanoylamino)-4,5,6,7-tetrahydrobenzo[b]-thiophene-3-carboxylic acid ethyl ester (2a). Colorless crystals, m.p. 54–55 °C; yield 86%; IR (cm-1): 3,230 (NH), 1,678 (CO, ester), 1,662 (CO, amide); 1H-NMR (CDCl3) δ: 1.36 (3H, t, J = 7.3 Hz, CH2CH3), 1.76 (4H, m, 2CH2), 1.86 (4H, m, 2CH2), 2.48 (2H, t, J = 7.0 Hz, CH2-CO), 2.61 (2H, t, J = 5.5 Hz, CH2), 2.74 (2H, t, J = 5.5 Hz, CH2), 3.54 (2H, t, J = 7 Hz, CH2Cl), 4.29 (2H, q, J = 7.3 Hz, CH2CH3), 11.01 (s,NH); 13C-NMR (CDCl3) δ: 22.61, 26.92, 26.44, 31.89 (4C, 4CH2), 23.05, 24.71, 35.90, 44.47 (4C, CH2) 14.38 (CH2CH3), 60.54 (CH2CH3), 111.46, 126.75, 130.78, 147.54 (4C, thiophene carbons), 166.75 (CO-NH), 169.32 (ester CO).
2-(5-Chloropentanamido)-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-3-carboxylic acid ethyl ester (2b). Colorless crystals, m.p. 50 °C; yield 86%; IR (cm–1): 3,363 (NH), 1,697 (CO, ester), 1,681 (CO, amide); 1H-NMR (CDCl3) δ: 1.35 (3H, t, J = 7 Hz, CH2CH3), 1.60 (4H, m, 2CH2), 1.83 (4H, m, 2CH2), 2.45 (2H, t, J = 6.9 Hz, CH2-CO), 2.67 (2H, m, CH2), 2.99 (2H, m, CH2), 3.52 (2H, t, J = 6.9 Hz, CH2Cl), 4.29 (2H, q, J = 7 Hz, CH2CH3), 11.18 (1H, s, NH); 13C-NMR (CDCl3) δ: 22.63, 26.99, 27.86, 31.87, 32.24 (5C, 5CH2), 28.31, 28.62, 35.89, 44.46 (4C, CH2) 14.38 (CH2CH3), 60.72 (CH2CH3), 112.82, 130.95, 136.36, 145.45 (4C, thiophene carbons), 169.29 (CO), 166.78 (CO-NH).
3.3. 2-[5-(4-(2-Methoxyphenyl)-1-piperazin-1-yl]pentanoylamino)-4,5,6,7-tetrahydrobenzo[b]-thio-phene-3-carboxylic acid ethyl ester (3a)
A mixture of 2a (3.9 mmol), 1-(2-methoxyphenyl)piperazine (750 mg, 3.9 mmol), and potassium carbonate (540 mg, 3.9 mmol) was dissolved in DMF (7 mL) and refluxed under stirring for 2 h. After cooling, the suspension was filtered and the solution was extracted with chloroform and washed with water. The organic layers were dried over anhydrous sodium sulfate and evaporated under reduced pressure. Recrystallization from ethanol gave white crystals, m.p. 105–106 °C; yield 94%; IR (cm–1): 3,261 (NH), 1,691 (CO, ester), 1,660 (CO, amide); 1H-NMR (CDCl3) δ: 1.34 (3H, t, J = 7.32 Hz, CH2CH3), 1.60 (2H, m, CH2), 1.75 (6H, m, 3 CH2), 2.43 (4H, t, J = 7.3 Hz, 2 CH2-N piperazine), 2.48 (4H, t, J = 7.3 Hz, 2 CH2-N piperazine), 2.61 (6H, br, 3 CH2), 2.73 (2H, br s, CH2-CO), 3.81 (3 H, s, O-CH3), 4.28 (2H, q, J = 7.3 Hz, CH2CH3), 6.80–6.97 (4H, m, Ar-H), 11.01 (s, NH); 13C-NMR (CDCl3) δ: 22.95, 23.07, 23.40, 24.42 (4C, 4 CH2 N-piperazine), 26.46, 36.80, 50.72, 53.55 (4C, 4 CH2), 55.40 (OCH3), 26.46 (2 C), 58.26 (2 C) (4C, 4 CH2), 14.41 (CH2CH3), 60.50 (CH2CH3), 111.30, 126.60, 130.71, 147.74 (4 C, thiophene carbons), 111.19, 118.22, 121.02, 122.88, 141.46, 152.32 (phenyl carbons), 166.75 (CO-NH), 169.32 (CO).
3.4. 2-(5-(4-(2-Methoxyphenyl)piperazin-1-yl)pentanamido)-5,6,7,8-tetrahydro-4H-cyclohepta[b]-thiophene-3-carboxylic acid ethyl ester (3b)
Compound 3b was prepared from equimolar amounts of 2b and 1-(2-methoxyphenyl)piperazine following the same conditions and work up described for the preparation of 3a. The product could not be induced to crystallize, although the NMR spectra proved the structure. Yield 85%; 1H-NMR (CDCl3) δ: 1.72 (4H, m, 2CH2), 1.84 (2H, m, CH2), 2.55 (2H, m, CH2), 2.85 (2H, m, CH2); 13C-NMR (CDCl3) δ: 19.64, 26.20, 28.62, 29.15 (2C) (cycloheptane carbons).
3.5. 3-Amino-2-[4-[4-(2-methoxyphenyl)-piperazin-1-yl]butyl]-5,6,7,8-tetrahydro-3H-benzo[4,5]-thieno[2,3-d]pyrimidin-4-one (4a)
A mixture of
3a (20 mmol) and hydrazine hydrate (4 mL, 80 mmol) was refluxed for 12 h in ethanol (10 mL). After cooling, the product was collected and washed with ethanol, dried and recrystallized from ethanol, to yield
4a colorless crystals, m.p. 163–164 °C; yield 92%; IR (cm
–1): 3,273 and 3,145 (NH), 1,672 (C=O);
1H and
13C-NMR data: see
Table 1; MS:
m/z 467 (M
+, 2.02%), 452 (M
+-CH
3), 18.27%), 204 (M
+-(NH
2, (CH
2)
4-N
2C
4H
8-C
6H
4-OCH
3)), 24.29%), 119 (M
+= [C
6H
4-N-CH
2CH
3]
+, 100%).
3.6. 3-Amino-2-[4-[4-(2-methoxyphenyl)-piperazin-1-yl]butyl]-6,7,8,9-tetrahydro-3H,5H-[4,5]thieno[2,3-d]pyrimidin-4-one (4b)
This compound was prepared from 3b following the same conditions and work up described for the preparation of 4a. Colorless crystals, m.p. 183–185 °C; yield 81%; IR (cm–1): 3310, 3135 (NH), 1670 (C=O); 1H and 13C-NMR data: almost the same data as 4a above except for an additional CH2 in the cycloheptane moiety in 4b: δH 1.79 (4H, m, 2 CH2), 1.95 (2H, m, CH2), 2.51 (2H, m, CH2), 2.87 (2H, m, CH2) and δC 19.81, 25.14, 27.40, 29.15 (2C, cycloheptane carbons); MS: m/z 481 (M+, 7.3%), 466 (M+ - CH3), 15.39%).
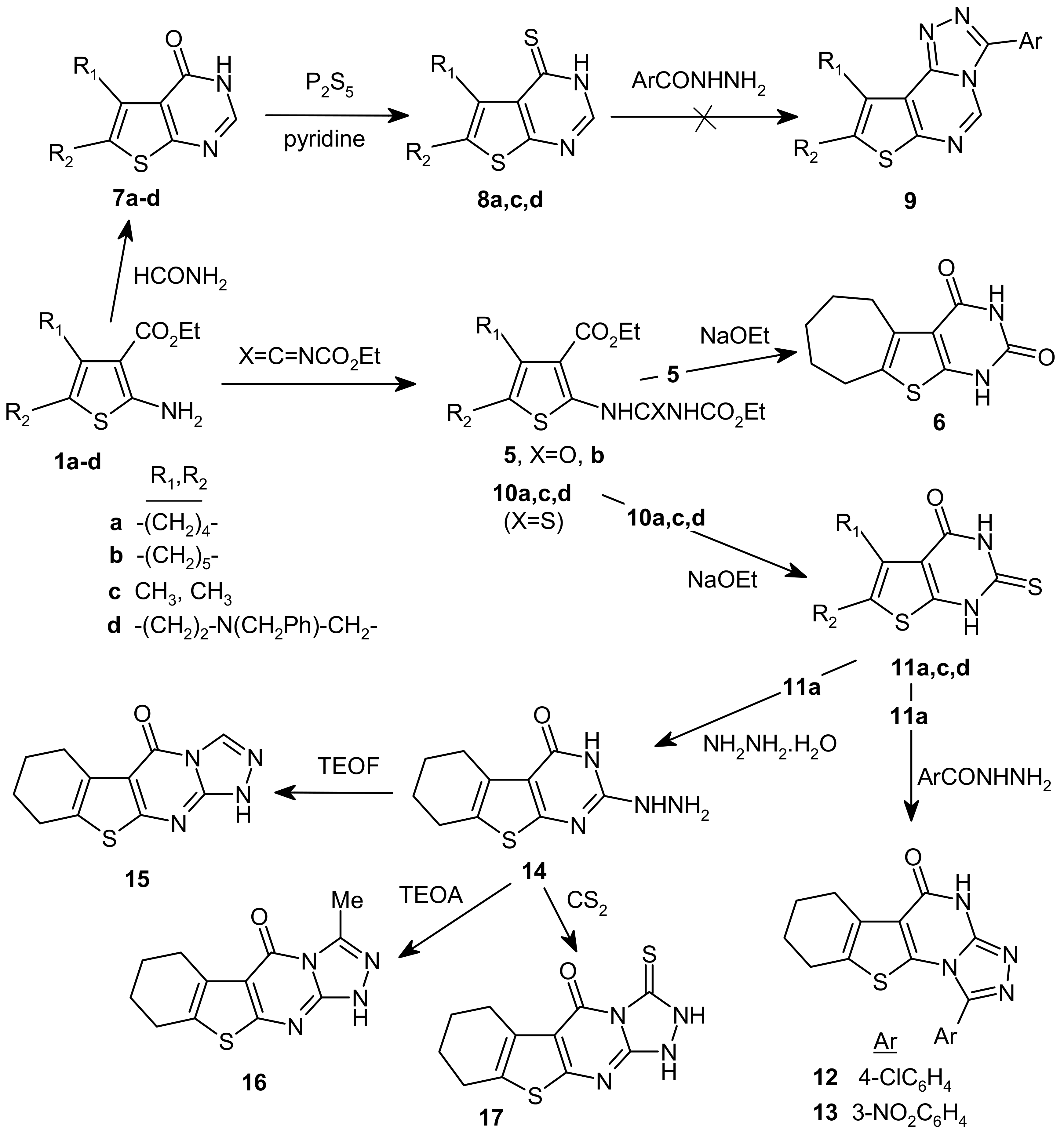
3.7. 2-(3′-Ethoxycarbonyluriedo)-3-ethoxycarbonyl-5,6,7,8-tetrahydro-4H-cyclohepta[b] thiophene (5)
A mixture of 1b (1.195 g, 5 mmol) and ethyl isocyanatoformate (575 mg, 5 mmol) was placed in a 50 mL conical flask covered with a funnel glass and then irradiated with microwaves (950 W) for 35 seconds. The cold reaction mixture was treated with ethanol and the solid product was filtered and recrystallized from ethanol-water to give colorless needles, m.p. 167–169 °C; yield 88%; IR (cm–1): 3,207, 3,115 (2 NH), 1,739 (CO, ester), 1,678, 1,675 (2 CO, amide); 1H-NMR (CDCl3) δ: 1.32 (3H, t, CH3), 1.36 (3H, t, CH3), 1.64 (4H, m, 2 CH2), 1.83 (2H, m, CH2), 2.71 (2H, t, CH2), 3.05 (2H, t, CH2), 4.30 and 4.40 (each 2H, q, O-CH2), 7.45 (1H, s, NH), 12.43 (1H, s, NH); 13C-NMR (CDCl3) δ: 14.34 and 14.44 (each 3H, t, CH3), 27.06, 27.89, 28.35, 28.69, 32.33 (5C-5CH2), 60.72, 62.98 (2C, O-CH2), 114.22, 131.20, 137.18, 144.16 (thiophene ring), 165.52 (CO), 153.11, 149.56 (2 CO-amide); MS: m/z 354 (M+, 89.5%), 265 (M+-NHCO2C2H5,H, 23%), 192 (M+-NHCO2C2H5,H, CO2C2H5, 100%), 164 (M+-NHCO2 C2H5, H, CO2C2H5, CO, 74%).
3.8. 1,5,6,7,8-Hexahydro-3H-cyclohepta[4,5]thienopyrimidin-2,4-dione (6)
Compound 5 (1770 mg, 5 mmol) was added to a solution of sodium ethoxide [sodium metal (120 mg), absolute ethanol (15 mL)], and refluxed 0.5 h, then the solvent was removed under reduced pressure. The residue was treated with water, then acidified with dil. HCl 1:1 (pH = 4), and the solid formed was collected and recrystallized from ethanol to give 6 as colorless crystals, m.p. > 300 °C; yield 82%; IR (cm–1): 3,095, 3,153 (2 NH), 1,708, 1,666 (2 CO), 2,600–3,300 (OH); 1H-NMR (DMSO-d6) δ: 1.53 (4H, m, 2CH2), 1.80 (2H, m, CH2), 2.69 (2H, t, CH2), 3.10 (2H, t, CH2), 10.95 (s, NH), 11.75 (NH); 13C-NMR (DMSO-d6) δ: 27.21, 27.31, 27.98, 28.87, 32.41 (5C, 5 CH2), 113.92, 129.56, 136.79, 149.83 (thiophene carbons), 150.87, 160.73 (2 CO); MS: m/z 236 (M+, 100%), 208 (M+-C2H4, 39.4%), 193 (M+-C2H4,NH), 36%), 165 (M+-C2H4,NH, CO), 48%), 137 (M+-C2H4, NH, 2CO), 42.4%), 133 (M+-(C2H4, NH, 2CO, H), 33.3%).
3.9. General procedure for synthesis of 5,6-disubstituted-3H-thieno[2,3-d] pyrimidin-4-ones 7a-d
A mixture of
1a-d (2 mmol) and formamide (20 mL) was heated under reflux for 1.5 h, then left to cool to room temperature overnight. The solid formed was filtered, washed with water, dried and recrystallized from ethanol [
13].
5,6,7,8-Tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (7a). Fine brown needles, m.p. 224–226 °C; yield 92%; IR (cm–1): 3,157 (NH), 3,007 (C-H), 1,658 (CO), 1,589 (C=C); 1H-NMR (DMSO-d6) δ: 1.76 (4H, m, 2 CH2), 2.71 (2H, t, CH2), 2.86 (2H, t, CH2), 7.98 (H-2), 12.29 (br, s, NH); 13C-NMR (DMSO-d6) δ: 22.31, 23.00, 24.98, 25.88 (aliphatic carbons), 123.23, 131.35, 132.64, 145.37 (4C, thiophene carbons), 158.21 (C=N), 162.96 (C=O); MS: m/z 206 (M+, 20%), 178 (M+-(HCNH), 1%), 57 (C2SH, 100%).
3,5,6,7,8,9-Hexahydrocyclohepta[4,5]thieno[2,3-d]pyrimidin-4-one (7b). Fine brown crystals, m.p. 118–220 °C; yield 90%; IR (cm–1): 3,151 (NH), 3,012 (C-H), 1,656 (CO), 1,598 (C=C); 1H-NMR (DMSO-d6) δ: 1.60 (4H, m, 2 CH2), 1.84 (2H, m, CH2), 2.82 (2H, m, CH2), 3.25 (2H, t, CH2), 7.98 (H-2), 12.30 (br, s, NH); 13C-NMR (DMSO-d6) δ: 27.70 (2C), 27.81, 29.57, 32.50 (aliphatic carbons), 123.78, 136.98, 145.07, 149.00 (4C, thiophene carbons), 158.75 (C=N), 161.40 (C=O); MS: m/z 220 (M+, 63%), 192 (M+-C2H4, 45%), 165 (M+-C2H4,HCN, 39%), 122 (M+-C2H4,HCN, CO, NH, 27%), 58, C2H2S, 100%).
5,6-Dimethylthieno[2,3-d]pyrimidin-4-one (7c). Fine yellow crystals, m.p. 269–270 °C; yield 93%; IR (cm–1): 3,151 (NH), 3,057 (C-H), 1,656 (CO), 1,558 (C=C); 1H-NMR (DMSO-d6) δ: 2.33 (3H, s, CH3), 2.37 (3H, s, CH3), 7.98 (1H, s, CH), 12.29 (br, s, NH); 13C-NMR (DMSO-d6) δ: 13.09, 13.40 (2C,CH3), 129.25, 129.79, 132.64, 145.23 (4C, thiophene carbons), 158.49 (C=N), 162.19 (C=O); MS: m/z 180 (M+, 100%), 165 (M+-CH3, 1%), 57 (C2SH, 94%).
7-Benzyl-5,6,7,8-tetrahydro-3H-pyrido[4′,3′:4,5]thieno[2,3-d]pyrimidin-4-one (7d). Fine yellow crystals, m.p. 234–235 °C; yield 89%; IR (cm–1): 3,157 (NH), 3,068 (C-H), 1,660 (CO), 1,581 (C=C); 1H-NMR (DMSO-d6) δ: 2.75 (2H, t, CH2), 2.93 (2H, t, CH2), 2.60 (2H, s, CH2), 2.69 (2H, s, CH2), 7.26–7.35 (5H, m, Ar-H), 8.02 (1H, s, CH), 12.39 (1H, br, s, NH); 13C-NMR: (DMSO-d6) δ: 26.20, 49.58, 51.51, 61.38 (4C, aliphatic carbons), 127.65, 128.85 (2C), 129.34 (2C), 129.80 (Ar-CH), 122.87, 130.38, 138.69, 145.74 (4C, thiophene carbons), 158.21 (C=N); 163.43 (C=O); Ms: m/z 297 (M+, 93%), 206 (M+-CH2-C6H5, 32%), 178 (M+-CH2-C6H5, HCN, H, 100%).
3.10. General procedure for synthesis of 4,5-disubstituted-3H-thieno[2,3-d] pyrimidin-4-thiones 8a,c,d
A mixture of compound 7a,c,d (10 mmol), phosphorous pentasulphide (4.02 g, 30 mmol) and dry pyridine (50 mL) was refluxed with stirring for 2 h. The mixture was evaporated to dryness under reduced pressure and the residue was boiled with water (100 mL) for one hour. After cooling overnight in refrigerator, the formed solid was recrystallized from a suitable solvent.
5,6,7,8-Tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-thione (8a). Recrystallized from ethanol give yellow crystals, m.p. 235–237 °C; yield 90%; IR (cm–1): 3,132 (NH), 3,061 (Ar-CH), 1,562 (C=C), 1,185 (C=S); 1H-NMR (DMSO-d6) δ: 1.61 (4H, m, 2CH2), 2.53 (2H, t, CH2), 3.04 (2H, t, CH2), 7.62 (1H, s, CH), 13.14 (br, s, NH); 13C-NMR (DMSO-d6) δ: 22.26, 22.40, 25,63, 27.99 (aliphatic carbons), 132.55, 132.91, 135.68, 142.42 (thiophene carbons); 161.18 (C=N); 178.24 (C=S); MS: m/z 222 (M+, 100%), 180 (M+- NCHNH, 66%), 149 (M+-NCHNH, S, +H, 66%).
5,6-Dimethyl-3H-thieno[2,3-d]pyrimidin-4-thione (8c). Light brown crystals, m.p. 247–248 °C (ethanol); yield 88%; IR (cm–1): 3,120 (NH), 3,068 (Ar-CH), 1,568 (C=C) 1,181 (C=S); 1H-NMR (CDCl3) δ: 2.32 (3H,s,CH3), 2.57 (3H, s, CH3), 8.09 (1H, s, CH), 13.56 (br, s, NH); 13C-NMR (CDCl3) δ: 13.05, 15.05 (2C, CH3), 130.29, 131.97 (2C), 143.13 (4C, thiophene carbons), 160.13 (C=N), 178.09 (C=S); MS: m/z 196 (M+, 100%), 181 (M+-CH3, 24%), 163 (M+-SH, 54%).
7-Benzyl-5,6,7,8-tetrahydro-3H-pyredo-[4′,3′:4,5]thieno[2,3-d]pyrimidin-4-thione (8d). Yellow powder, m.p. 204–207 °C (ethanol-DMF); yield: 50%; IR (cm–1): 3,415 (NH), 3,028 (Ar-CH), 1,180 (C=S), 1,629 (C=C); 1H-NMR (DMSO-d6) δ: 2.98 (2H, CH2), 3.37 (2H, CH2), 4.13 (2H, CH2), 4.31 (2H, s, CH2-Ph), 7.23–7.39 (5H, m, Ar-H), 8,32 (H-2), 13.78 (br.s, NH).
3.11. General procedure for the synthesis of 10a,c,d
A mixture of 1a,c,d (10 mmol) and ethoxycarbonyl isothiocyanate (10 mmol) in ethanol (5 mL) was placed in a 50 mL conical flask covered with a funnel glass and then irradiated with microwaves (950 W) for 35–40 seconds. The cold reaction mixture was treated with ethanol and the solid product was filtered and recrystallized from a suitable solvent.
2-(Ethoxycarbonylamino-carbothioyl)amino-4,5,6,7-tetrahydrobenzo[4,5]thiophene- 3-carboxylic acid ethyl ester (10a). Yellow crystals, m.p. 193–195 °C (ethanol); yield 92%; IR (cm–1): 3,182 (NH), 1,732 (ester CO), 1,680 (amide CO), 1,562, 1,533 (C=C), 1,147 (C=S), 1,201 (C-O); 1H-NMR (CDCl3) δ: 1.30 and 1.34 (each 3H, t, J = 7.3 Hz, CH3CH2), 4.32 and 4.40 (each 2H, q, J = 7.3 Hz, CH2CH3), 1.76 (4H, m, 2CH2), 2.63 (2H, t, CH2), 2.77 (2H, t, CH2), 8.19 (br. s, NH), 14.03 (s, NH); 13C-NMR (CDCl3) δ: 14.36, 14.44 (2C, 2 CH3), 22,92 (2C, 2 CH2), 24.45, 26.49, 60.81, 63.09, 116.09, 128.31, 131.78, 147.16 (thiophene carbons), 151.41 (CO-N), 165.66 (CO), 173.27 (C=S).
2-(Ethoxycarbonylamino-carbothioyl)amino-4,5-dimethyl[4,5]thiophene-3-carboxylic acid ethyl ester (10c). Yellow crystals, m.p. 274–176 °C (ethanol); yield 90%; IR (cm–1): 3,176 (NH), 1,728 (C=O), 1,683 (amide C=O), 1,564, 1,531 (C=C), 1,170 (C=S), 1,293 (C-O); 1H-NMR (CDCl3) δ: 1.32 and 137 (each 3H, t, CH3CH2), 2.64 (3H, s, CH3-C4), 2.69 (3H, s, CH3-C5), 4.43 (2H, q, CH2-CH3), 4.32 (2H, q, CH2-CH3), 8.07 (IH, s, NH), 14.03 (H, s, NH); 13C-NMR (CDCl3) δ: 12.46, 14.36, 14.41, 14.57 (4C, 4CH3), 60.93, 63.12 (2C, 2CH2), 117.24, 125.01, 130.30, 146.23 (4C, thiophene carbons) 151.40 (CO), 165.68 (CO), 173.16 (C=S).
2-(Ethoxycarbonylamino-carbothioyl)amino-7-benzyl-5,6,7,8-tetrahydro-3H-pyredo-[4′,3′:4,5]thio-phene-3-carboxylic acid ethyl ester (10d). Light yellow powder, m.p. 205–207 °C (ethanol and DMF); yield 91%; IR (cm–1): 3,064 (NH), 3,024, 1,728 (ester CO), 1,683 (amide CO), 1,564, 1,525 (C=C), 1,110 (C=S), 1,213 (C-O); 1H-NMR (DMSO-d6) δ: 1.33 (3H, t, J = 7.4 Hz,CH3), 1.36 (3H, t, J = 7.4 Hz, CH3), 4.35 (2H, q, J = 7.4 Hz, CH2), 4.43 (2H, q, J = 7.4 Hz, CH2), 2.79 (2H, m, CH2), 2.94 (2H, m, CH2), 3.57 (2H, s, CH2), 3.71 (2H, s, CH2), 8.14 (NH-CO), 14.09 (NH); 13C-NMR (DMSO-d6) δ: 14.37 (CH3), 14.44 (CH3), 60.92 and 63.19 (2CH2-O), 27.01, 50.32, 51.36, 62.16 (CH2-N), 115.61, 125.48, 130.35, 147.91 (4C, thiophene carbons), 127.37, 128.46 (2C), 129.24 (2C), 138.12, 151.42 (amide CO), 165.52 (ester CO), 173.36 (C=S).
3.12. General procedure for the synthesis of 11a,c,d
These susbstances were prepared according to a method reported in the literature [
23]. Compound
10 (1 mmol) was dissolved in solution of sodium ethoxide (230 mg sodium and 15 mL of absolute ethanol) and the solution was heated under reflux for 30 min. The solvent was then evaporated under vacuum, some water was added to the residue, and the pH of the mixture was adjusted to 4 with hydrochloric acid. The product that separated was collected and crystallized from a suitable solvent.
2-Thioxo-2,3,5,6,7,8-hexahydro-1H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (11a). White powder, m.p. 275–277 °C (ethanol); yield 88%; IR (cm–1): 3,157 (NH), 1,672 (CO), 1,577, 1,556 (C=C), 1,157 (C=S); 1H-NMR (DMSO-d6) δ: 1.78 (4H, m, 2CH2), 2.70 (2H, m, CH2), 2.80 (2H, m, CH2), 12.34 (s, NH), 13.36 (s, NH); 13C-NMR (DMSO-d6) δ: 22.13, 23.08, 24.50, 25.47 (4C, 4 CH2), 117.04, 128.78, 131.43, 150.54 (4C, thiophene carbons), 157.58 (CO), 173.40 (C=S);
5,6-Dimethyl-2-thioxo-2,3-dihydro-1H-thieno[2,3-d]pyrimidin-4-one (11c). Light yellow powder, m.p. 205–207 °C (ethanol); yield 82%; IR (cm–1): 3,427, 3,404 (NH), 1,664 (CO), 1,604, 1,556 (C=C), 1,174 (C=S); 1H-NMR (DMSO-d6) δ: 2.20 (3H, s, CH3), 2.24 (3H, s, CH3), 12.26 (s, NH), 13.26 (s, NH); 13C-NMR (DMSO-d6) δ: 12.49, 13.01 (2C, 2CH3), 117.71, 125.68, 129.21, 149.79 (4C, thiophene carbons), 157.71 (CO), 173.16 (C=S).
7-Benzyl-2-thioxo-2,3,5,6,7,8-hexahydro-1H-pyrido[4′,3′:4,5]thieno[2,3-d]pyrimidin-4-one (11d). Orange powder, m.p. >300 °C (ethanol and DMF); yield 20%; IR (cm–1): 3,350, 3,180 (NH), 3,076, 1,681 (CO), 1,544, 1,519 (C=C), 1,130 (C=S); 1H-NMR (DMSO-d6) δ: 3.15 (2H, CH2), 3.39 (2H, CH2), 4.24 (2H, CH2), 4.43 (2H, s, CH2-Ph), 7.26-7.38 (5H, m, Ar-H), 11.40 (br. s, NH), 12.50 (s, NH).
3.13. Synthesis of 12 and 13
To a solution of 11a (1.19 g, 5 mmol) was added aroyl hydrazine (5 mmol) in n-butanol (15 mL) and the mixture was heated under reflux for 20 h. The solid obtained was cooled, collected and recrystallized from butanol.
1-(4-Chlorophenyl)-6,7,8,9-tetrahydro-4H-benzo[4,5]thieno[2,3-d][1,2,4]triazolo[3,4-b]-pyrimidin-5-one (12): Fine yellow crystals, m.p. 218–219 °C; yield 86%; IR (cm–1): 3,253 (NH), 3,035 (Ar-CH), 1,656 (CO), 1,600 (C=N), 1,566 (C=C); 1H-NMR (DMSO-d6) δ: 1.70 (4H, 2 CH2), 2.63 (2H, m, CH2), 2.73 (2H, m, CH2), 7.52 (2H, d, J = 8.8 Hz, C6H4), 7.82 (2H, d, J = 8.8 Hz, C6H4), 9.87 (br. s, NH); 13C-NMR (DMSO-d6) δ: 22.05, 22.00, 24.42, 25.39 (aliphatic carbons), 116.97, 128.70, 131.36, 150.56 (4C, thiophene carbons), 128.96 (2C), 129.41 (2C), 136.43 (C-Cl), 132.56 (C, Ar-C1), 157.82 (C=N), 165.34 (C=N), 173.31 (C=O); MS: m/z 356 (M+, 1.1%), 220 (M+-Cl-C6H4-CN) + H, 9%), 193 ( M+-Cl-C6H4-CN, C2H4 + 2H, 13%).
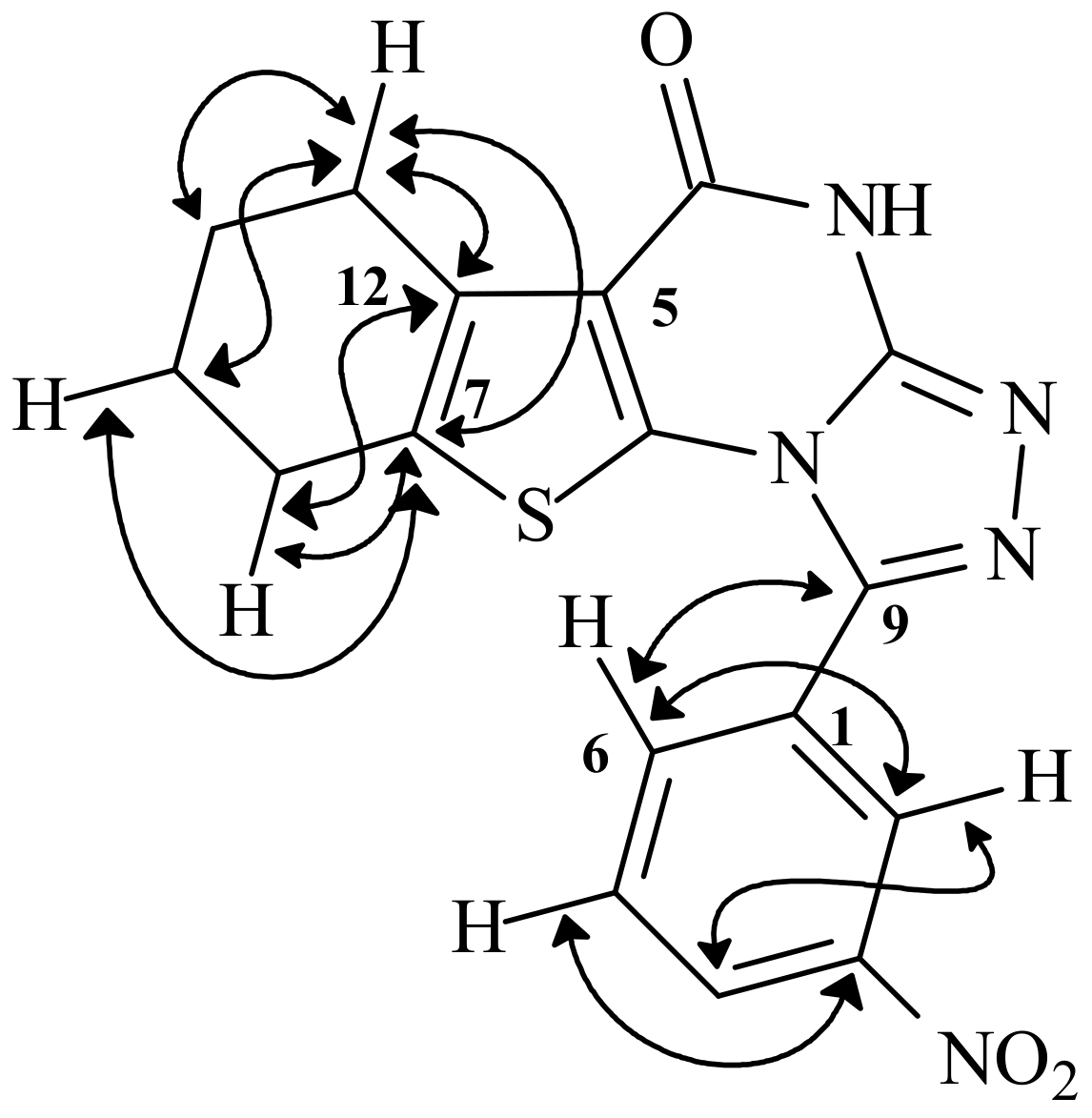
1-(3-Nitrophenyl)-6,7,8,9-tetrahydro-4H-benzo[4,5]thieno[2,3-d][1,2,4]triazolo[3,4-b]-pyrimidin-5-one (13): Fine yellow powder, m.p. 234–235 °C; Yield 79%; IR (cm–1): 3,251 (NH), 3,107–3,041, 1,687 (CO), 1,650 (C=N), 1,579 (C=C), 1,523, 1,346 (N=O); 1H-NMR (DMSO-d6) δ: 1.70 (4H, 2CH2), 2.60 (2H, m, CH2), 2.71 (2H, m, CH2), 7.73 (1H, t, J = 8 Hz, Ar-5CH-m), 8.22 (1H, d, J = 6.0 Hz, Ar-6CH-o), 8.33 (1H, d, J = 6.0 Hz, Ar-4CH-p), 8.60 (1H, s, Ar-2CH-o), 12.20 (s, NH); 13C-NMR (DMSO-d6) δ: 21.24, 22.37, 23.78, 24,73 (4C, aliphatic carbons), 116.36, 128.01, 130.77, 150.00 (4C, thiophene carbons), 121.66, 125.45, 129.97, 133.10 (4C, Ar-CH), 134.71 (C), 147.78 (C-NO2), 156.81 (C=N), 163.47 (C=N), 172.86 (C=O); MS: m/z 367 (M+, 87%), 221 (M+- (N=C-C6H3-NO2 )+2H, 24%), 207 ((M+- [(N=C-C6H3-NO2 ) + 2H]-NH2, 20%).
3.14. 2-Hydrazino-5,6,7,8-tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (14)
A mixture of
11a (950 mg, 4 mmol) and 99% hydrazine hydrate (4 mL, 80 mmol) in pyridine (20 mL) was heated under reflux for 15 h. The mixture was evaporated under reduced pressure and the residue was treated with ethanol. The solid product was filtered and washed several times with ethanol to give colorless crystals, m.p. 265–267 °C; yield 83%; IR (cm
–1): 3,319, 3,265 (NH, NH
2), 1,658 (CO), 1,597 (C=C);
1H-NMR (DMSO-d
6) δ: 1.73 (4H, m, 2CH
2), 2.50 (2H, m, CH
2), 2.75 (2H, m, CH
2), 8.20 (NH), NH
2 not observed;
13C-NMR (DMSO-d
6) δ:22.49, 23.30, 24.73, 25.84 (aliphatic carbons), 114.25, 125.12, 130.60, 155.67 (thiophene carbons), 158.36 (C=N), 167.14 (CO) [
13].
3.15. 5,6,7,8-Tetrahydro-1H-benzo[4,5]thieno[2,3-d][1,2,4]triazolo[3,4-a]-pyrimidin-4-one (15)
A mixture of 14 (1.18 g, 5 mmol) and excess triethyl orthoformate (10 mL) was heated under reflux with stirring for 2 h. The excess of the orthoformate was removed under reduced pressure. The formed solid was collected, washed with ethanol, dried and recrystallized from ethanol to give a yellow powder, m.p. 277–279 °C; yield 84%; IR (cm–1): 3,118 (NH), 3,039, 1,672 (CO), 1,608 (C=N), 1,554 (C=C); 1H-NMR (DMSO-d6): δ 1.77 (4H, 2CH2), 2.62 (2H, CH2),2.83 (2H, CH2), 9.01 (1H, s, CH), 14.04 (1H, s, NH); 13C-NMR (DMSO-d6) δ: 22.40, 23.17, 24,91, 25.94 (4C, aliphatic carbons), 112.08, 126.64, 130.19, 148.87 (4C, thiophene carbons), 132.50 (CH, triazole), 151.93 (C=N), 167.82 (C=O); MS: m/z : 246 (M+, 87%), 245 (M+-H, 43%), 195(M+-HCN, C2H4 + 4H, 37%).
3.16. 3-Methyl-5,6,7,8-tetrahydro-1H,2H-benzo[4,5]thieno[2,3-d][1,2,4]triazolo[3,4-a]-pyrimidin-4-one (16)
A mixture of 14 (1.18 g, 5 mmol) and excess triethyl orthoacetate (10 mL) was heated under reflux with stirring for 2 h. The solvent was removed under reduced pressure, The formed solid was collected washed with ethanol, dried and recrystallized from ethanol, give a white powder, m.p. 292–294 °C; yield 79%; IR (cm–1); 3,232 (NH), 1,714 (CO), 1,701 (C=N), 1,620 (C=C); 1H-NMR (DMSO-d6): δ 2.98 (3H, s, CH3), 1.68 (4H, 2CH2), 2.68 (2H, CH2), 2.76 (2H, CH2), 12.30 (1H, s, NH); MS m/z: 260 (M+, 100%), 245 (M+-CH3, 15%), 232 (M+-(C2H4), 57%).
3.17. 3-Thioxo-5,6,7,8-tetrahydro-1H,2H-benzo[4,5]thieno[2,3-d][1,2,4]triazolo[3,4-a]-pyrimidin-4-one (17)
A mixture of 14 (1.18 g, 5 mmol) in pyridine (15 ml) and carbon disulfide (380 mg, 5 mmol) was heated under reflux for 5h. After cooling, the obtained solid was collected and recrystallized from acetic acid to give a white powder, m.p. >300 °C; yield 81%; IR (cm–1): 3,105 (br, NH), 2,945, 1,664 (CO); 1H-NMR (DMSO-d6) δ: 1.75 (4H, 2CH2), 2.73 (2H, br, CH2), 2.83 (2H, br, CH2), 12.57 (br. s, NH), 13.86 (br. s, NH); 13C-NMR (DMSO-d6) δ: 22.19, 22.92, 24.41, 25.16 (4C, aliphatic carbons), 118.33, 130.93, 131.72, 139.32 (4C, thiophene carbons), 144.98 (C=N), 157.71 (C=O), 158.81 (C=S); MS: m/z 278 (M+, 100%), 250 ( M+-C2H4, 43%), 245 (M+-SH , 31%).
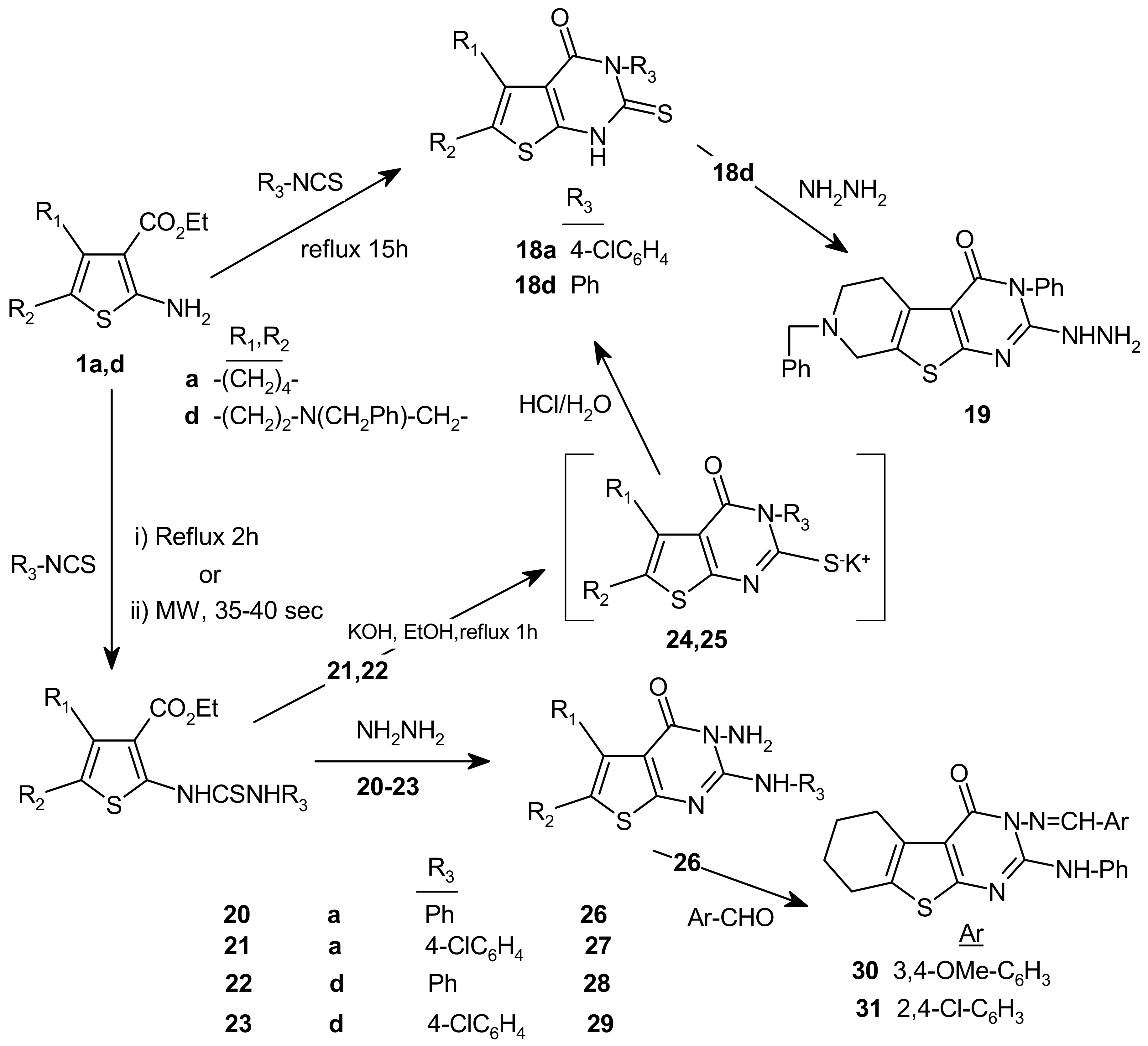
3.18. Synthesis of disubstituted thienyl-2-thioureides 20-23
3.18.1. Method A
2-Amino-3-ethoxycarbonyl thiophene
1a,d (100 mmol) was dissolved in hot ethanol (100 mL) and phenyl (or
p-clorophenyl) isothiocyanate (110 mmol) was added dropwise with stirring. The reaction mixture was heated under reflux on water bath for 2 h, then left to cool overnight and the separated crude solid was filtered, washed with ethanol, and recrystallized from ethanol [
12].
3.18.2. Method-B
A mixture of
1a,d (20 mmol) and phenyl (or
p-clorophenyl) isothiocyanate (20 mmol) placed in a 50 mL conical flask covered with a funnel glass and then irradiated with microwaves (950 W) for 35–40 seconds. The cold reaction mixture was then treated with ethanol and the solid product was filtered off and recrystallized from ethanol [
13].
2-(3-Phenylthioureido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid ethyl ester (20). Fine colorless needles, m.p. 187–189 °C; yield: (78%)A, (89%)B; IR (cm–1): 3,196 (NH), 3,032 (Ar-CH), 1,656 (CO), 1,554 (C=C), 1,195 (C=S); 1H-NMR (CDCl3) δ: 0.97 (3H, J = 7.3 Hz, t, CH2CH3), 1.43 (4H, m, 2CH2), 2.27 (2H, m, CH2), 2.41 (2H, m, CH2), 3.87 (2H, J = 7.3 Hz, q, CH2-CH3), 6.89 (t, J = 7.32 Hz, Ar-CH(p)), 7.05 (2H, t, J = 7.32 Hz, Ar-CH(m)), 7.11 (2H, d, J = 8.08 Hz, Ar-CH(o)), 10.03 (s, NH), 11.70 (s, NH); 13C-NMR (CDCl3) δ: 14.25 (CH3), 22.92, 23.02, 24,09, 26.17 (aliphatic carbons); 60.14 (O-CH2), 112.01, 125.89, 130.35, 150.17 (4C, thiophene carbons); 124.51, 125.89(2C), 128.94 (2C), 137.81 (aromatic carbons), 166.13 (C=O); 175.99 (C=S); MS: m/z : 360 (M+, 20%), 326 (M+-SH, H, 17%), 225 (M+- (SH, H, C6H5, CN) + 2H, 33%).
2-(3-(4-Chlorophenyl)thioureido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic acid ethyl ester (21). Yellow needles, m.p. 198–199 °C; yield (71%)A, (88%)B; IR (cm–1): 3,141 (NH), 3,078 (Ar-CH), 1,652 (CO), 1,558 (C=C), 1,195 (C=S); 1H-NMR (CDCl3) δ: 1.25 (3H, t, CH2CH3), 1.75 (4H, m, 2CH2), 2.60 (2H, m, CH2), 2.70 (2H, m, CH2), 4.15 (2H, q, CH2CH3), 7.28 (2H, d, J = 8.8 Hz, Ar-CH), 7.41 (2, d, J = 8.8 Hz, Ar-CH), 7.92 (br. s, NH), 12.25 (1H, s, NH); 13C-NMR (CDCl3) δ: 14.12 (CH3), 22.92, 23.02, 24.40, 26.38 (4C, aliphatic carbons), 60.65 (O-CH2), 113.23, 127.28, 133.18, 149.81 (thiophene carbons), 126.80 (2C), 130.13 (2C), 130.96, 134.66 (phenyl carbons), 166.68 (C=O), 176.06 (C=S); MS: m/z 394 (M+, 26%) 396 (M++2, 15%), 225 (M+-(OCH2CH3,NH-C6H4Cl) + 2H, 39%), 179 (M+-(OCH2CH3,NHCSNH-C6H4Cl), 100%).
6-Benzyl-2-(3-phenylthioureido)-4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxylic acid ethyl ester (22). Orange needles, m.p. 274–276 °C; yield (64%)A,(77%)B; IR (cm–1): 3,196 (br, NH), 3,032 (Ar-CH), 1,656 (CO), 1,554 (C=C), 1,197 (C=S); 1H-NMR (CDCl3) δ: 1.21 (3H, J = 7.3 Hz, t, CH2CH3), 2.93 (4H, m, 2CH2), 3.37 (4H, br. s, 2CH2), 4.12 (2H, J = 7.3 Hz, q, CH2CH3), 7.32 (5H, m, C6H5), 7.46 (5H, m, C6H5), 7.95 (s, NH), 12.11 (s, NH); MS: m/z 450 (M+-H, 16%), 91 ((Ph-CH2), 100%).
6-Benzyl-2-(3-(4-chlorophenyl)thioureido)-4,5,6,7-tetrahydro thieno [2,3-c]pyridine-3-carboxylic acid ethyl ester (23). Yellow needles, m.p. 259–261 °C; yield (54%)A, (69%)B; IR (cm–1): 3,176 (NH), 3,053, 3,031 (Ar-CH), 1,652 (CO), 1,558 (C=C), 1,197 (C=S); 1H-NMR (CDCl3) δ: 1.24 (3H, t, J = 7.36 Hz, CH2CH3), 2.85 (4H, m, 2CH2), 3.57 (2H, s, CH2), 3.74 (2H, s, CH2), 4.17 (2H, q, J = 7.36 Hz, CH2CH3), 7.22 (2H, d, J = 8.8 Hz, Ar-CH), 7.38 (2H, d, J = 8.8 Hz, Ar-CH), 7.24–7.39 (5H, m, C6H5), 8.37 (br. s, NH), 12.15 (s, NH); 13C-NMR (CDCl3) δ: 14.25 (CH3), 26.46, 50.13, 51.02, 61.85 (4C, CH2); 60.77 (O-CH2), 112.56, 127.12, 133.19, 150.65 (4C, thiophene ring); 123.90, 126.85 (2C), 130.09 (2C), 134.62 (C6H5), 126.87 (2C), 128.52 (2C), 129.17, 129.41 (p-Cl-C6H4), 166.35 (C=O), 176.03 (C=S); MS: m/z 485 (M+, 0.02%), 316 (M+-(NH-C6H4Cl,OCH2CH3) +H, 9%), 168 (M+- (NH-C6H4Cl,OCH2CH3, CH2C6H5, NCH2,CO), 80%).
3.19. General procedure for the preparation of the monopotassium salts of 24 and 25
A mixture of the compounds
21,
22 (13.5 mmol) and potassium hydroxide (760 mg, 13.5 mmol) in absolute ethanol (55 mL) was heated under reflux with stirring for 1h. The suspension was filtered while hot, and the solid was washed with hot absolute ethanol to give
24) [
13] and
25 which were both used without any further purification.
3.20. General procedure for the preparation of 18a,d
3.20.1. Method A
A suspension of potassium salts of 24, 25 in water (50 mL) was acidified with concentrated hydrochloric acid and stirred at room temperature for 30 min. The solid was collected by filtration, washed with water and recrystallized from the suitable solvent to give 18a,d.
3.20.2. Method B: Synthesis of 18d
A mixture of 1d (3.16 g, 10 mmol) and phenyl isothiocyanate (1350 mg, 10 mmol) in acetonitrile (30 mL) was heated under reflux for 15 h in the presence of anhydrous potassium carbonate (1.4 g). The reaction mixture was then cooled, filtered, diluted with water (10 mL) and neutralized with 2M hydrochloric acid. The product obtained was filtered, washed with water, dried and recrystallized from acetic acid.
Monopotassium salt of 3-(4-chlorophenyl)-2-thioxo-2,3,5,6,7,8-hexahydro-1H-[4,5]thieno[2,3-d]-pyrimidin-4-one (24) and its 2-thioxo derivative 18a. Yields 54% (24) and 72% (18a), recrystallized from ethanol to give a white powder, m.p. > 300 °C; IR (cm–1): 3,130 (NH), 3,060, 3,039, 1,693 (CO), 1,524, 1,490 (C=C); 1H-NMR (CDCl3) δ: 1.85 (4H, m, 2CH2), 2.75 (2H, m, CH2), 2.91 (2H, m, CH2), 7.34 (2H, d, J = 8.8 Hz, Ar-CH), 7.55 (2H, d, J = 8.8 Hz, Ar-CH), 7.26 (1H, s, NH); MS: m/z 348 (M+, 50%), 206 (M+-C6H5Cl, SH + 2H, 58%), 111 (M+- (C6H5Cl, SH,2C2H4, N,+ 3H), 56%).
Monopotassium salt of 7-benzyl-3-phenyl-2-thioxo-2,3,5,6,7,8-hexahydro-1H-pyrido[3′,4′:4,5]- thieno[2,3-d]-pyrimidin-4-one (25) and its 2-thioxo derivative 18d. Yields: 49% (25) and 59%A, 46%B (18d); Yellow powder, m.p. 273–274°C (acetic acid); IR (cm–1): 3,390 (NH), 3,061, 1,689 (CO), 1,523 (C=C); MS: m/z 405 (M+, 27%), 314 (M+-(CH2-Ph), 7%), 286 (M+-(CH2-Ph, N-CH2), 29%).
3.21. 7-Benzyl-3-phenyl-2-hydrazino-5,6,7,8-tetrahydro-1H-pyrido[3′,4′:4,5]thieno[2,3-d]-pyrimidin-4-one (19)
A mixture of
18d (1.62 g, 4 mmol) and 99% hydrazine hydrate (4 mL, 80 mmol) in pyridine (20 mL) was heated under reflux for 15 h. The mixture was evaporated under reduced pressure and the residue was treated with ethanol. The solid product was filtered, washed with ethanol, dried and recrystallized from ethanol [
13] to give an orange powder, m.p. 186–187 °C; Yield 75%; IR (cm
–1): 3,311–3,145 (NH, NH
2), 3,034, 1,689 (C=O), 1,546, 1,504 (C=C);
1H-NMR (CDCl
3) δ: 2.82 (2H, m, CH
2), 2.99 (2H, m, CH
2), 3.61 (2H, s, CH
2), 3.72 (2H, s, CH
2), 3.89 (2H, s, NH
2), 5.40 (s, NH), 7.22 (2H, d,
J = 8.8 Hz, Ar-CH), 7.38 (2H, d,
J = 8.8 Hz, Ar-CH), 7.23–7.58 (6H, m, C
6H
5);
13C-NMR (CDCl
3) δ: 25.68, 49.97, 51.61, 62.10 (4C, CH
2), 115.39, 125.48, 133.65, 152.68 (4C, thiophene carbons), [127.36, 128.46 (2C), 128.78 (2C), 129.24 (2C), 129.95, 130.14, 130.69 (2C), 138.14 (12C, 2C6H4)], 158.41 (C=N), 164.85 (C=O); MS:
m/z 403 (M
+,47%), 387 (M
+-(NH
2), 20%), 312 (M
+-CH
2-Ph, 8%), 284 (M
+-CH
2-Ph , N-CH
2, 66%).
3.22. General procedure for the preparation of 26–29
A mixture of thiouredo derivatives 20–23 (10 mmol) and hydrazine hydrate (20 mmol) in ethanol (100 mL) was heated under reflux for 3–4 h. The solid that separated upon cooling was filtered, washed with water, dried and recrystallized from ethanol.
3-Amino-2-phenylamino-5,6,7,8-tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (26). Fine colorless crystals, m.p. 204–205 °C; Yield 56%; IR (cm–1); 3,323, 3,309, 3,259 (NH2, NH), 3,034, 1,672 (CO), 1,606, 1,544 (C=C); 1H-NMR (DMSO-d6) δ: 1.76 (4H, m, 2CH2), 2.63 (2H, m, CH2), 2.82 (2H, m, CH2), 5.59 (2H, s, NH2), 7.06 (1H, t, J = 7.3 Hz, Ar- CH(p)), 7.34 (2H, t, J = 7.3 Hz, Ar-CH(m)), 7.74 (2H, d, J = 8.08 Hz, Ar-CH(o)), 9.35 (1H, s, NH); 13C-NMR (DMSO-d6) δ: 22.42, 23.24, 24.84, 25.77 (aliphatic carbons), 115.19, 127.48, 130.72, 149.30 (thiophene carbons), 121.39, 123.69, 129.14, 138.83 (phenyl carbons), 157.99 (C=N), 166.13 (C=O); MS: m/z 312 (M+, 20%), 297 (M+-NH2)+ H, 21%), 235 (M+- Ph, 36%), 221 (M+-NH Ph + H, 46%).
3-Amino-2-(4-chlorophenyl)amino-5,6,7,8-tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (27). colorless crystals, m.p. 166–167 °C; Yield: 46%; IR (cm–1); 3,334, 3,330, 3,203 (NH2, NH), 3,099, 1,664 (CO), 1,587, 1,546 (C=C); 1H-NMR (DMSO-d6) δ: 1.78 (4H, m, 2CH2), 2.64 (2H, m, CH2), 2.87 (2H, m, CH2), 4.57 (2H, s, NH2), 7.29 (2H, d, J = 8.8 Hz, Ar-CH), 7.59 (2H, d, J = 8.8 Hz, Ar-CH), 8.53 (s, NH); 13C-NMR (DMSO-d6) δ: 22.34, 23.11, 25.00, 25.45 (4C, aliphatic ring), 115.54, 128.56, 130.69, 147.15 (4C, thiophene ring); 121.14 (2C), 129.05 (2C), 129.15, 136.41 (aromatic carbons), 158.20 (C=N), 163.09 (C=O); MS: m/z 346 (M+, 98%), 348 (M++1, 54%), 312 (M+-Cl + H, 14%), 330 (M+-NH2, 15%), 331 (M+-NH2 + H, 6%), 220 (M+-NH2, Ph-Cl) +H, 5%), 206 (M+-NH2, NH Ph-Cl) + 2H, 38%), 179 (M+-NH2, NH Ph-Cl, HCN + 2H, 32%).
7-Benzyl-3-Amino-2-phenylamino-5,6,7,8-tetrahydro-3H-pyrido[3′4′:4,5]thieno[2,3-d]pyrimidin-4-one (28). Light yellow crystals, m.p. 150–152 °C; Yield 32%; IR (cm–1): 3,317, 3,300 (NH2, NH), 3,032 (Ar-CH), 1,691 (CO), 1,589, 1,543 (C=C); 1H-NMR (CDCl3) δ: 2.78 (2H, t, CH2), 2.99 (2H, t, CH2), 3.46 (2H, s, CH2), 3.68 (2H, s, CH2), 4.81 (2H, s, NH2), 7.07 (1H, t, J = 7.3 Hz, Ar-CH(p)), 7.34 (2H, t, J = 8.08 Hz, Ar-CH(m)), 7.55 (2H, d, J = 8.08 Hz, Ar-CH(o)), 7.40–7.35 (5H, m, C6H5), 8.42 (1H, s, NH); 13C-NMR (CDCl3) δ: 25.78, 50.21, 51.60, 62.52 (aliphatic carbons), 114.73, 127.48, 137.73, 147.52 (4C, thiophene carbons), [120.02 (2C), 123.63, 125.61, 128.51, 129.02 (2C), 129.44 (2C), 129.65 (2C), 137.84, aromatic carbons], 157.97 (C=N), 163.66 (C=O); MS : m/z 403 (M+,49%), 387 (M+(-NH2 ),14%), 311 (M+-(C6H5, NH),16%), 284 (M+-(CH2C6H5, N-CH2, 43%), 176(M+-(CH2C6H5, N-CH2, NH2, NH-C6H5), 9%), 150 (M+-(CH2C6H5, N-CH2, NH2, NH-C6H5, HCN) + (H), 23%).
7-Benzyl-3-Amino-2-(4-chlorophenyl)amino-5,6,7,8-tetrahydro-3H-pyrido[3′4′:4,5]thieno[2,3-d]pyrimidin-4-one (29). Light yellow crystals, m.p. 192–194 °C; Yield: 29%; IR (cm–1): 3,450–3,313 (br, NH2), 3,201 (NH), 3,032 (Ar-CH), 1,674 (CO), 1,537 (C=C); 1H-NMR (CDCl3) δ: 2.78 (2H, m, CH2), 3.02 (2H, m, CH2), 3.44 (2H, s, CH2), 3.68 (2H, s, CH2), 4.86 (2H, s, NH2), 7.25 (2H, d, J = 8.8 Hz, Ar-CH), 7.47 (2H, d, J = 8.8 Hz, Ar-CH), 7.37–7.30 (5H, m, C6H5), 8.40 (s, NH); 13C-NMR (CDCl3) δ: 25.78, 50.31, 51.56, 62.62 (4C, 4CH2), 114.88, 127.55, 137.72, 147.25 (4C, thiophene carbons); [121.12 (2C), 125.83, 128.55 (2C), 128.99 (2C), 129.02 (2C), 129.51 (2C), 136.30, 12C, 2C6H5], 157.81 (C=N), 163.35 (C=O); MS : m/z 437 (M+, 88%), 421 (M+ - NH2 , 18%), 345 (M+-[CH2C6H5] + H, 29%), 318 (M+-CH2C6H5,HCN + H, 68%), 302 (M+- CH2C6H5, HCN,-NH2, +H), 21%), 91 (CH2C6H5, 100%).
3.23. Synthesis of 30, 31
A mixture of 26 (1.56 g, 5 mmol) and aromatic aldehyde (5 mmol) in acetic acid (30 mL) was heated under reflux for 4 h. Then the mixture was cooled and the solid separated was filtered, dried and recrystallized from a suitable solvent.
3-[N′-(3,4-Dimethoxybenzylidene)-hydrazino]-2-phenylamino-5,6,7,8-tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (30). Light yellow crystals, m.p. 192–194 °C; Yield 56%; IR (cm–1): 3,325 (NH), 3,053, 3,007, 1,672 (CO), 1,600, 1,570 (C=C); MS: m/z 460 (M+, 24%), 297 (M+-(N=CHC6H3 (OMe)2) + H, 44%), 180 (M+-(N=CHC6H3 (OMe)2, NHC6H5, HCN) + 2H, 16%).
3-[N′-(2,4-Dichlorobenzylidene)-hydrazino]-2-phenylamino-5,6,7,8-tetrahydro-3H-benzo[4,5]thieno[2,3-d]pyrimidin-4-one (31). Yellow powder, m.p. 192–194 °C; Yield 51%; IR (cm–1): 3,319 (NH), 3,068, 1,668 (CO), 1,598, 1,537, 1,544 (C=C); MS: m/z 468 (M+, 57%), 470( M+ +2, 33%), 323 (M+- (C6H3Cl2), 29% ), 268 (M+-(C6H3Cl2,HCN, C2H4), 96% ), 178 (M+-(C6H3Cl2, HCN, C2H4, NHC6H5 + 2H, 46% ).
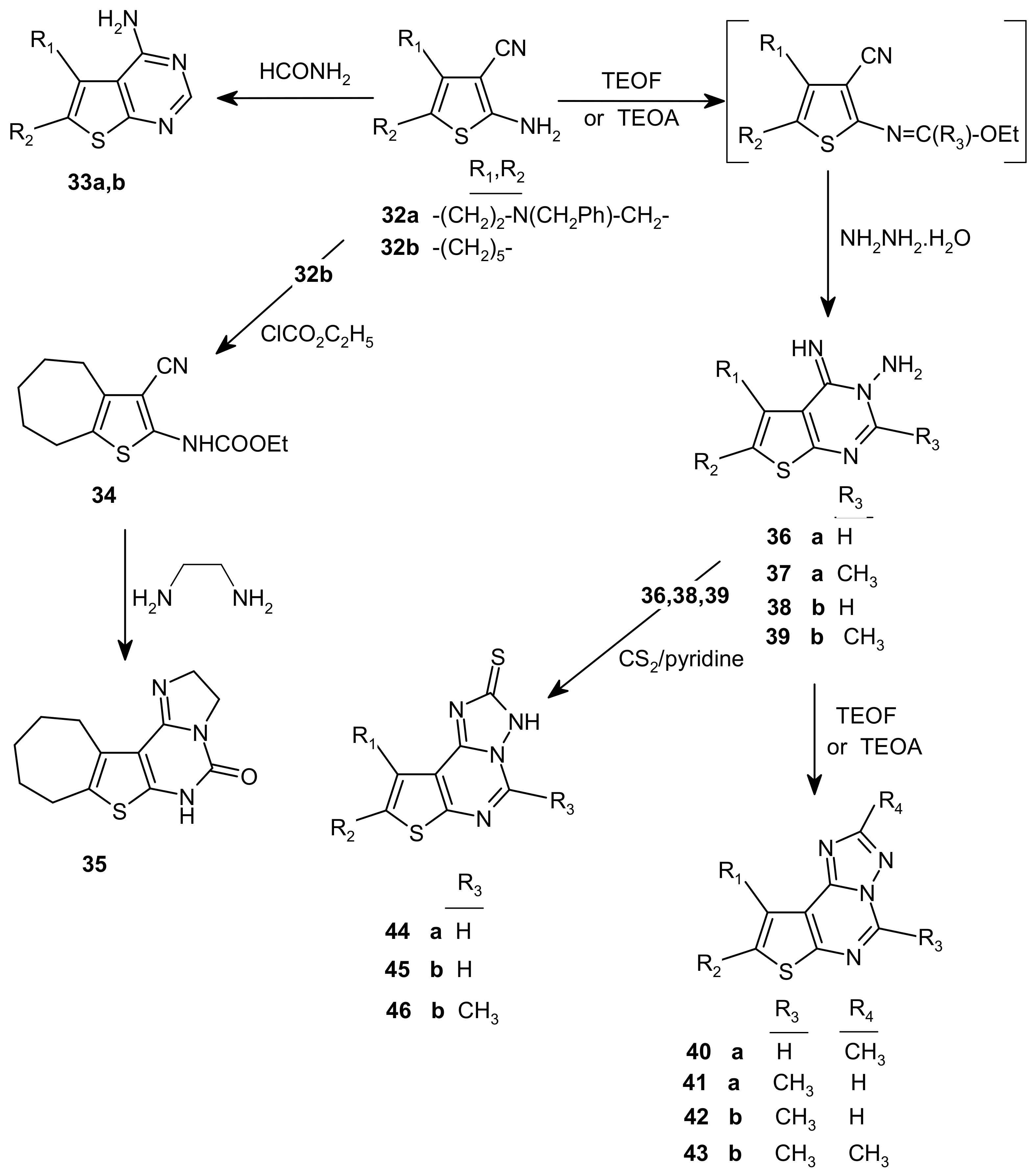
3.24. Synthesis of 4-amino-5,6-disubstituted[4,5]thieno[2,3-d]pyrimidines 33a,b
A mixture of compound 32a,b (10 mmol) and formamide (10 mL) was heated under reflux for 2 h, then left to cool overnight at ambient temperature. The solid formed was filtered, dried and recrystallized from ethanol [24].
7-Benzyl-4-amino-5,6,7,8-tetrahydropyrido[3′,4′:4,5]thieno[2,3-d]pyrimidine (33a). Light brown crystals, m.p. 236–237 °C; yield: 73%; IR (cm–1): 3,413, 3,313 (NH2), 3,050, 1,633, 1,552 (C=C); 1H-NMR (DMSO-d6) δ: 2.77 (2H, t, CH2), 2.96 (2H, t, CH2), 3.62 (2H, s, CH2), 3.70 (2H, s, CH2), 6.85 (2H, s, NH2), 7.27–7.36 (5H, m, C6H5), 8.18 (1H, s, H-2); 13C-NMR (DMSO-d6) δ: 26.17, 49.43, 51.93, 61.16 (4C, 4CH2), 115.02, 127.68, 138.71, 153.70 (thiophene carbons), 126.13, 128.85 (2C), 129.13, 129.37, (2C) (C6H5), 158.72 (C-2), 166.30 (C-4); MS : m/z 296 (M+, 25%), 205 (M+-(CH2-C6H5), 50%), 177 (M+-(CH2-C6H5,NCH2), 63%), 162 (M+- ( CH2-C6H5,NCH2, NH2 ) + H, 2%).
4-Amino-6,7,8,9-tetrahydro-5H-cyclohepta[4,5]thieno[2,3-d]pyrimidine (33b). Violet crystals, m.p. 264–266 °C; Yield: 66%; IR (cm–1): 3,352, 3,327 (NH2), 3,143, 1,651, 1,556 (C=C); 1H-NMR (DMSO-d6) δ: 1.65 (4H, m, 2CH2), 1.83 (2H, m, CH2) 2.83 (2H, t, CH2), 3.00 (2H, t, CH2), 6.92 (2H, s, NH2), 8.15 (1H, s, H-2); 13C-NMR (DMSO-d6) δ: 26.98, 27.40, 29.11 (2C), 31.34 (5C, 5 CH2), 116.66, 132.68, 135.55, 152.96 (4C, thiophene carbons), 158.70 (C-2), 161.79 (C-4); MS: m/z 219 (M+, 100%), 204 (M+-(NH2) + H, 23%), 190 (M+-(NH2, CH2) + H, 52%).
3.25. (3-Cyano-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophene-2-yl)-carbamic acid ethyl ester (34)
A mixture of 32b (3.84 g, 20 mmol) and ethyl chloroformate (2.3 mL, 24 mmol) in pyridine (40 mL), was stirred for 30 h at room temperature, the solvent was removed under reduced pressure to obtain a dark oil, treated with 3–5 mL of water, the the precipitate collected by filtration, dried and recrystallized from ethanol to give violet crystals of 34, m.p. 135–137 °C; Yield: 49%; IR (cm–1): 3,215 (NH), 2,223 (CN), 1,724 (CO), 1,571, 1,556 (C=C), 1,240 (C-O); 1H-NMR (CDCl3) δ: 1.33 (3H, t, J = 7.32 Hz, CH3), 1.64 (4H, m, 2CH2), 1.82 (2H, m, CH2), 2.67 (4H, m, 2CH2), 4.26 (2H, q, J = 7.32 Hz, CH2-O), 7.69 (1H, s, NH); 13C-NMR (CDCl3) δ: 14.44 (CH3), 27.34, 28.06, 29.13, 29.22, 32.04 (5C, 5CH2), 62.87 (CH2-O), 95.30, 130.98, 136.04, 146.13 (4C, thiophene carbons), 114.81 (CN), 152.67 (C=O).
3.26. 2,3,5,7,8,9,10,11-Octahydro-[1,3]-imidazolo[2,3-c]-4-oxo-cyclohepta[4,5]thieno[2,3-d]pyrimidine (35)
Ethylenediamine (1 mL) was added dropwise with stirring to a solution of compound 34 (264 mg, 1 mmol) in DMF (5 mL) within 2 h at 150 °C. After cooling, water (20 mL) was added to the solution and the precipitated crystals were collected by filtration, washed with water, dried and crystallized from ethanol to give colorless crystals, m.p. 275–276 °C; yield: 80%; IR (cm–1): 3,138 (NH), 1,643 (CO), 1,573, 1,525 (C=C); 1H-NMR (CDCl3) δ: 1.52 (4H, m, 2CH2), 1.69 (2H, m, CH2), 2.53 (2H, t, J = 5.8 Hz, CH2), 2.94 (2H, t, J = 5.8 Hz, CH2), 3.79 (2H, m, N-CH2), 3.90 (2H, m, N-CH2), 2.44 (1H, s, NH); 13C-NMR (CDCl3) δ: 24.17, 24.85, 25.10, 26.31, 29.09 (5C, 5CH2), 40.04 (2C, 2CH2-N), 112.00, 121.50, 127.35, 132.11 (thiophene carbons), 146.21 (CN), 149.52 (C=O); MS: m/z 261 (M+, 100%), 220 (M+-(NCH2CH2) + H, 50%).
3.27. 7-Benzyl-4-imino-3-amino-5,6,7,8-tetrahydro-4H-pyrido[3′,4′:4,5] thieno [2,3-d] pyrimidine (36)
A mixture of 32a (20 mmol) and triethylorthoformate (20 mL) was heated under reflux for 4 h, and the excess of the reagent was then removed under vacuum, A mixture of hydrazine hydrate (99%, 5 mL) and ethanol (15 mL) was added to the residue with stirring, and it was allowed to stand overnight at room temperature. The solid obtained was filtered, washed with ethanol, dried and crystallized from ethanol. Fine brown crystals, m.p. 166–167 °C; Yield: 67%; IR (cm–1); 3,354, 3,253, 3,174 (NH2, NH), 3,094–3,031 (Ar-CH), 1,652, 1,606 (C=N), 1,554, 1,541 (C=C); 1H-NMR (CDCl3) δ: 2.98 (2H,t,CH2), 3.21 (2H, s, CH2), 3.72 (4H, m, 2 CH2), 5.42 (2H, s, NH2), 7.25-7.37 (5H, m, C6H5), 8.08 (H, s, CH), 8.87 (1H, s, NH).
7-Benzyl-4-imino-3-amino-2-methyl-5,6,7,8-tetrahydro-4H-pyrido[3′,4′:4,5] thieno[2,3-d] pyrimidine (37). Prepared from 32a (20 mmol) and triethyl orthoacetate (20 mL) following the same procedure as for 36 above. Fine light brown crystals, m.p. 172–174 °C; Yield : 54%; IR (cm–1): 3,330, 3,284, 3,178 (NH2, NH), 3,094–3,028 (Ar-CH), 1,652, 1,610 (C=N), 1,544, 1,521 (C=C); 1H-NMR (CDCl3) δ: 2.79 (3H, s, CH3), 2.83 (2H, t, J = 5.8 Hz, CH2), 2.97 (2H, t, J = 5.8 Hz, CH2), 3.58 (2H, s, CH2), 3.69 (2H, s, CH2), 6.14 (2H, br, s, NH2), 7.34–7.28 (6H, m, NH, C6H5); 13C-NMR (CDCl3) δ: 22.75 (CH3), 26.18, 49.43, 51.70, 61.94 (4C, 4CH2), 115.59, 127.18, 137.53, 154.59 (thiophene carbons), 127.50, 128.57 (2C), 128.18 (2C), 133.00 (6C, C6H5), 156.05 (C-2), 160.50 (C-4).
4-Imino-3-amino-6,7,8,9-tetrahydro-4H,5H-cyclohepta[4,5]thieno[2,3-d]pyrimidine (38). Prepared from 32b (20 mmol) with triethyl orthoformate (20 mL) following the procedure above described for 36. Fine colorless crystals, m.p: 164–165 °C; yield: 56%; IR (cm–1): 3,294, 3,286, 3,163 (NH2, NH), 3,053 (Ar-CH), 1,639, 1,614 (C=N), 1,562 (C=C); 1H NMR (CDCl3) δ: 1.79 (4H, m, 2CH2), 1.91 (2H, m, CH2), 2.74 (2H, m, CH2), 3.06 (2H, m, CH2), 4.75 (2H, brs, NH2), NH not observed, 7.96 (1H, s, C2-H); 13C-NMR (CDCl3) δ: 25.53, 27.34, 29.23, 29.36, 31.20 (5C, 5 CH2), 120.44, 135.02, 138.08, 146.41 (4C, thiophene carbons), 155.31 (C-2), 155.99 (C-4).
4-Imino-3-amino-2-methyl-6,7,8,9-tetrahydro-4H,5H-cyclohepta[4,5]thieno[2,3-d] pyrimidine (39). Prepared from 32b (20 mmol) with triethyl orthoacetate (20 mL) following the procedure of 36. Fine colorless crystals, m.p. 186–188 °C; Yield: 70%; IR (cm–1); 3,354, 3,284, 3,190 (NH2, NH), 3,001 (Ar-CH), 1,647, 1,606 (C=N), 1,552, 1,521 (C=C); 1H-NMR (CDCl3) δ: 1.71 (4H, m, 2CH2), 1.83 (2H, m, CH2), 2.67 (3H, s, CH3), 2.77 (2H, m, CH2), 3.00 (2H, m, CH2), 5.50 (2H, br, s, NH2), NH not observed; 13C-NMR (CDCl3) δ: 22.53 (1C, CH3), 26.41, 27.21, 29.14, 29.25, 30.96 (5C, 5 CH2), 117.51, 134.13, 138.23, 154.94 (thiophene ring), 155.55 (C-2), 157.28 (C-4).
3.28. 8-Benzyl-2-methyl-7,8,9,10-tetrahydropyrido[4′,3′-4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine (40)
Compound 36 (10 mmol) was heated under reflux for 2 h with an excess of triethyl orthoacetate(20 mL) and the excess of the reagent was then removed under vacuum. The solid obtained was washed with ethanol, dried and crystallized from ethanol to give brown crystals, m.p. 157–159 °C; Yield: 45%; IR (cm–1): 3,041 (Ar-CH), 1,618 (C=N), 1,556, 1,508 (C=C); 1H-NMR (CDCl3) δ: 2.65 (3H, s, CH3), 2.99 (2H, m, CH2), 3.27 (2H, m, CH2), 3.79 (4H, s, 2CH2), 7.34–7.38 (5H, m, C6H5), 9.06 (1H, s, CH); 13C-NMR (CDCl3) δ: 14.78 (CH3), 25.62, 49.79, 51.77, 61.91 (4C, 4 CH2), 119.65, 127.43, 136.20, 137.79 (thiophene carbons), 127.52, 128.55 (2C), 129.12 (2C), 135.44 (6C, C6H5), 149.29 (C=N), 154.06 (C=N), 165.25 (C=N); MS : m/z 335 (M+, 11%), 244 (M+-(CH2-C6H5), 10%), 216 (M+-(CH2-C6H5, NCH2), 37%).
8-Benzyl-4-methyl-7,8,9,10-tetrahydro-pyrido[4′,3′-4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine (41). Prepared from 37 (10 mmol) and triethyl orthoformate (20 mL) following the procedure given for 40. Fine light yellow scales, m.p. 142–144 °C; Yield 39%; IR (cm–1); 3,084–3,060 (Ar-CH), 1,652, 1,622 (C=N), 1,558, 1,517 (C=C); 1H-NMR (DMSO-d6) δ: 2.97 (3H, s, CH3), 3.04 (2H, m, CH2), 3.30 (2H, br, s, CH2), 3.82 (4H, m, 2CH2), 7.26-7.40 (5H, m, C6H5), 8.36 (1H,s,CH); MS: m/z 335 (M+, 8%), 244 (M+-(CH2-C6H5), 30%), 216(M+-(CH2-C6H5, NCH2), 100%).
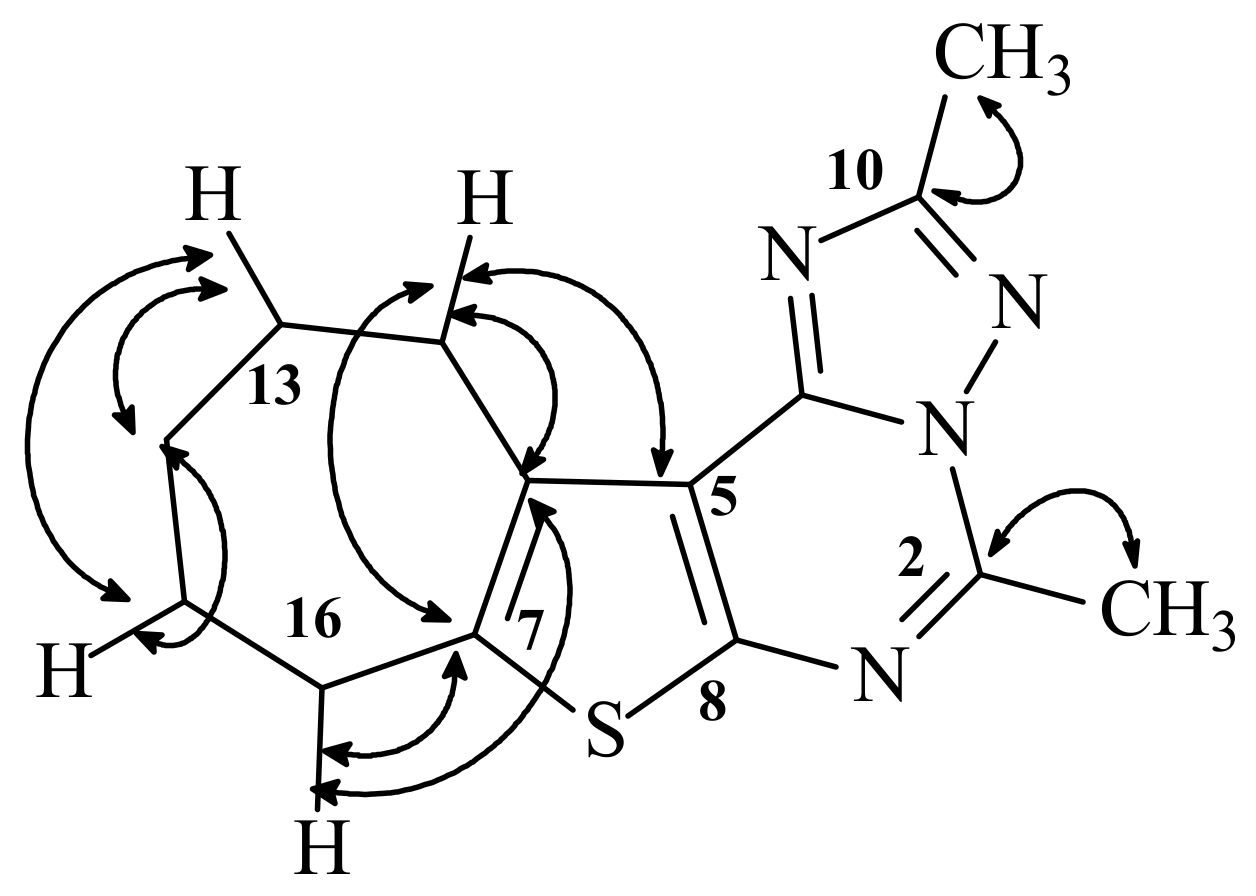
4-Methyl-8,9,10,11-tetrahydro-7H-cyclohepta[4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine (42). Prepared from 39 (10 mmol) and triethyl orthoformate (20 mL) following the procedure given above for 40. Fine colorless crystals, m.p. 159–160 °C; Yield: 67%; IR (cm–1): 3,089 (Ar-CH), 1,618 (C=N), 1,550, 1,521 (C=C); 1H-NMR (CDCl3) δ: 1.74 (4H, m, 2 CH2), 1.92 (2H, m, CH2), 2.91 (2H, m, CH2), 2.95 (3H, s, CH3), 3,41 (2H, m, CH2), 8.34 (1H, s, C-H); 13C-NMR (CDCl3) δ: 19.64 (CH3), 27.34, 27.85, 28.41, 30.46,32.42 (5C, 5CH2), 120.55, 134.11, 141.56, 145.43 (4C, thiophene carbons), 149.04 (C=N), 152.01 (C=N), 153.32 (C=N); MS: m/z 258 (M+, 100%), 243 (M+-(CH3), 37%), 230 (M+-(C2H4), 69%), 216 (M+-(C2H4 , CH3) + H, 18%).
4,2-Dimethyl-8,9,10,11-tetrahydro-7H-cyclohepta[4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine (43). Prepared from 39 (10 mmol) and triethyl orthoacetate (20 mL) following the procedure described above for 40, Fine colorless crystals, m.p. 189–190 °C; Yield: 64%; IR (cm–1): 2,922–2,854 (aliphatic CH), 1,618 (C=N), 1,550, 1,519 (C=C). 1H-NMR (CDCl3) δ: 1.77 (4H, m, 2 CH2), 1.93 (2H, m, CH2), 2.64 (3H, s, CH3), 2.94 (5H, CH2, CH3), 3,42 (2H, m, CH2). 13C-NMR (CDCl3) δ: 14.63 (CH3), 19.64 (CH3), 27.3, 27.8, 28.37, 30.4, 32.1 (5C, 5CH2), 119.89, 133.89, 140.71, 151.96 (thiophene carbons), 149.51 (C-4), 144.88 (C-10), 163.47 (C-2); MS: m/z 272 (M+, 82%), 243 (M+-(2CH3)+H, 60%), 230(M+-(CH3,C2H4)+H, 47%).
3.29. 8-Benzyl-7-8,9,10-tetrahydro-3H-pyrido[4′,3′:4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine-2-thione (44)
A mixture of 36 (1 mmol) in pyridine (10 mL) and carbon disulfide (400 mg, 5 mmol) was heated under reflux for 3 h. The solid obtained after cooling, was collected and recrystallized from ethanol. Fine orange scales, m.p. 237–238 °C; Yield: 36%; IR (cm–1): 3,379 (NH) 3,028 (Ar-CH), 2,850–2,920 (aliphatic CH), 1,525, 1,454 (C=C), 1,392 (C=S); 1H-NMR (CDCl3) δ:2.87 (2H, m, CH2), 3.68 (2H, m, CH2), 4.01 (4H, m, 2CH2), 7.50–7.52 (3H, m), 7.41–7.68 (2H, m), 8.14 (1H, s, H-2).
3.30. 8,9,10,11-Tetrahydro-3H,7H-cyclohepta[4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine-2-thione (45)
A mixture of 38 (1 mmol) in pyridine (10 mL) and carbon disulfide (400 mg, 5 mmol) was heated under reflux for 3 h. Work up following the procedure given above for 44 and recrystallization from ethanol gave 45 as yellow scales, m.p. 275–276 °C; Yield: 75%; IR (cm–1): 3,057 (Ar-CH), 2,9845–2,999 (aliphatic CH), 1,517, 1,454 (C=C), 1,365 (C=S). 1H-NMR (CDCl3) δ: 1.70–2.0 (6H, m, 3 CH2), 2.98 (4H, m, 2 CH2), 9.43 (1H, s, NH); 13C-NMR (CDCl3) δ: 26.56, 26.96, 28.99, 30.65, 31.38 (5C, 5CH2), 125.21, 132.25, 149.53, 153.5 (4C, thiophene carbons), 135.65 (C-2), 157.67 (C-4), 182.39 (C=S).
4-Methyl-8,9,10,11-tetrahydro-3H,7H-cyclohepta[4,5]thieno[3,2-e][1,2,4]triazolo[2,3-c]pyrimidine-2-thione (46). Prepared from 38 (1 mmol) in pyridine (10 mL) and carbon disulfide (400 mg, 5 mmol) following procedure described for 45. Yellow scales, m.p. 288–290 °C; Yield: 80%; IR (cm–1): 2,916, 2,846 (aliphatic CH2), 1,618 (C=N), 1,550, 1,519 (C=C); 1H-NMR (CDCl3) δ: 1.77 (4H, m, 2 CH2), 1.93 (2H, m, CH2), 2.91 (2H, m, CH2), 2.98 (2H, m, CH2), 3.03 (3H, s, CH3); 13C-NMR (CDCl3) δ: 22.37 (1C, CH3), 26.52, 26.96, 28.57, 30.35, 31.24 (5C, 5 CH2), 123.90, 131.68, 146.95, 153.68 (4C, thiophene carbons), 153.68 (C-2), 157.94 (C-4), 181.21 (C=S).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
