Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions

Abstract
:1. Introduction
2. Results and Discussion
2.1. Diastereoselective Ugi Reaction of 4-(Dimethylamino)-2-pyridinecarboxaldehyde (1)


| Entry | Concentration (M) | Yield (%) a | D.r. b |
|---|---|---|---|
| 1 | 0.1 | 79 | 55:45 |
| 2 | 0.2 | 84 | 62:38 |
| 3 | 0.5 | 98 | 63:37 |
| 4 | 1.0 | 91 | 60:40 |

| Entry | α-Amino acid | Product | Yield (%) a | D.r. b |
|---|---|---|---|---|
| 1 | L-Valine | 2a | 98 | 63:37 |
| 2 | L- t-Leucine | 2b | 70 | 62:38 |
| 3 | L-Isoleucine | 2c | 54 | 60:40 |
| 4 | L-Phenylalanine | 2d | 57 | 55:45 |
| 5 | L-Phenylglycine | 2e | 59 | 55:45 |
| 6 | L-Threonine | 2f | 77 | 50:50 |
| 7 | L-Serine | 2g | 85 | 53:47 |
| 8 | L-Proline | 2h | 19 | 51:49 |
2.2. Diastereoselective Ugi Reaction of 4-(Dimethylamino)-3-pyridinecarboxaldehyde (3)

| Entry | Temperature (°C) | Conversion (%) a | Yield (%) b | D.r. a |
|---|---|---|---|---|
| 1 | 20 | 50 | 37 | 84:16 |
| 2 | 40 | 79 | 53 | 90:10 |
| 3 | 50 | 92 | 57 | 89:11 |
| 4 | 60 | 97 | 48 | 85:15 |

| Entry | X | Y | Conversion (%) a | Yield (%) b | D.r. a |
|---|---|---|---|---|---|
| 1 | 1.1 | 1.1 | 91 | 54 | 91:9 |
| 2 | 1.3 | 1.1 | 94 | 55 | 89:11 |
| 3 | 1.5 | 1.1 | 97 | 55 | 89:11 |
| 4 | 1.1 | 1.3 | 96 | 59 | 89:11 |
| 5 | 1.1 | 1.5 | 95 | 57 | 85:15 |

{kind=link}
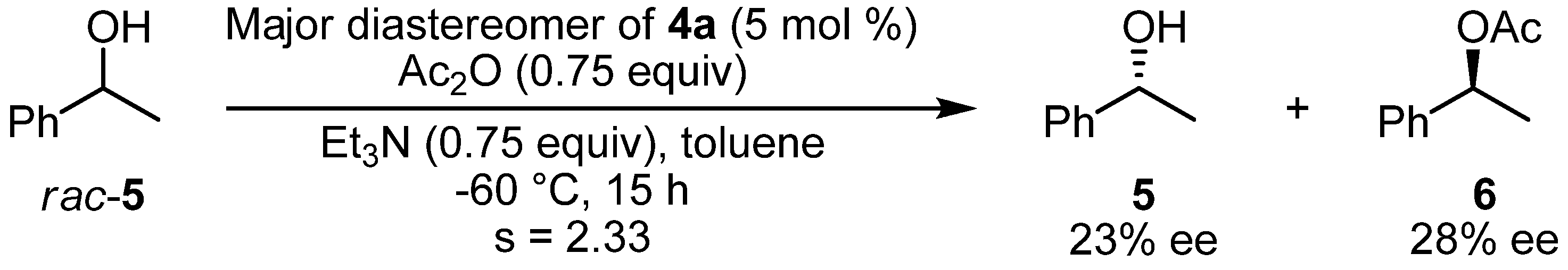{kind=link}
| Entry | α-Amino acid | Product | Yield a | D.r. b |
|---|---|---|---|---|
| 1 | L-Valine | 4a | 55 | 92:8 |
| 2 c | L-Valine | 4a | 57 | 88:12 |
| 3 d | L-Valine | 4a | 46 | 85:15 |
| 4 | L-Leucine | 4b | 63 | 86:14 |
| 5 | L- t-Leucine | 4c | 52 | 89:11 |
| 6 | L-Isoleucine | 4d | 55 | 93:7 |
| 7 | L-Phenylalanine | 4e | 60 | 88:12 |
| 8 | L-Phenylglycine | 4f | 45 | 59:41 |
| 9 | L-Serine | 4g | 43 | 77:23 |
| 10 | L-Methionine | 4h | 58 | 83:17 |
| 11 | O-t-Butyl L-threonine | 4i | 63 | 82:18 |
| 12 | γ-Methyl L-glutamate | 4j | 47 | 83:17 |
| 13 | 4-Benzyl L-aspartate | 4k | 52 | 79:21 |
| 14 | O-Benzyl L-serine | 4l | 54 | 75:25 |
| 15 | L-Histidine | 4m | 44 | 77:23 |
| 16 e | N-Benzyl L-valine | 4n | 18 | 92:8 |
| 17 | L-Proline | 4o | 51 | 74:26 |


2.3. Kinetic Resolution of a Racemic Alcohol by Chiral DMAP Derivatives

3. Experimental
3.1. General
3.2. General Procedure for the Ugi Reaction of 4-(Dimethylamino)-2-pyridinecarboxaldehyde (1)
Procedure for the Synthesis of 2h
3.3. General Procedure for the Ugi Reaction of 4-(Dimethylamino)-3-pyridinecarboxaldehyde (3)
3.4. Kinetic Resolution of a Racemic Alcohol by Chiral DMAP Derivative
3.5 X-ray Structure Report for (S,R)-4a
3.5.1. Data Collection
| Empirical formula | C19 H32 N4 O3 | |
| Formula weight | 364.49 | |
| Crystal Color, Habit | colorless, needle | |
| Temperature | 93(2) K | |
| Wavelength | 0.71075 Å | |
| Crystal system | orthorhombic | |
| Space group | P21212 (#18) | |
| Unit cell dimensions | a = 17.791(6) Å | a = 90° |
| b = 17.955(6) Å | b = 90° | |
| c = 6.363(2) Å | g = 90° | |
| Volume | 2032.6(11) Å3 | |
| Z | 4 | |
| Density (calculated) | 1.191 Mg/m3 | |
| Absorption coefficient | 0.082 mm−1 | |
| F(000) | 792 | |
| Crystal size | 0.60 × 0.08 × 0.06 mm3 | |
| Theta range for data collection | 3.22 to 27.49°. | |
| Index ranges | −23 ≤ h ≤ 23, −23 ≤ k ≤ 23, −8 ≤ l ≤ 8 | |
| Reflections collected | 31451 | |
| Independent reflections | 4665 [R(int) = 0.0603] | |
| Completeness to theta = 27.49° | 99.60% | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data / restraints / parameters | 4665 / 816 / 487 | |
| Goodness-of-fit on F2 | 0.966 | |
| Final R indices [I > 2sigma(I)] | R1 = 0.0471, wR2 = 0.1109 | |
| R indices (all data) | R1 = 0.0619, wR2 = 0.1202 | |
| Absolute structure parameter | 0.1(12) | |
| Extinction coefficient | 0.024(3) | |
| Largest diff. peak and hole | 0.228 and −0.238 e.Å−3 |
3.5.2. Data Reduction
3.5.3. Structure Solution and Refinement
4. Conclusions
Acknowledgements
References and Notes
- Zhu, J.; Bienaymé, H. Muticomponent Reactions; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar]
- Dömling, A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. [Google Scholar] [CrossRef]
- El Kaim, L.; Grimaud, L. Beyond the Ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Ramón, D.J.; Yus, M. Asymmetric multicomponent reactions (AMCRs): The new frontier. Angew. Chem. Int. Ed. 2005, 44, 1602–1634. [Google Scholar] [CrossRef]
- Kelly, C.L.; Lawrie, K.W.M.; Morgan, P.; Willis, C.L. Ugi four component condensations using aldehydes with an asymmetric centre at C-2. Tetrahedron Lett. 2000, 41, 8001–8005. [Google Scholar]
- Ahmadian, H.; Nielsen, B.; Bräuner-Osborne, H.; Johansen, T.N.; Stensbøl, T.B.; Sløk, F.A.; Sekiyama, N.; Nakanishi, S.; Krogsgaard-Larsen, P.; Madsen, U. (S)-Homo-AMPA, a specific agonist at the mGlu6 subtype of metabotropic glutamic acid receptors. J. Med. Chem. 1997, 40, 3700–3705. [Google Scholar]
- Madsen, U.; Frydenvang, K.; Ebert, B.; Johansen, T.N.; Brehm, L.; Krogsgaard-Larsen, P. N-Methyl-D-aspartic acid receptor agonists: Resolution, absolute stereochemistry, and pharmacology of the enantiomers of 2-amino-2-(3-hydroxy-5-methyl-4-isoxazolyl)acetic acid. J. Med. Chem. 1996, 39, 183–190. [Google Scholar] [CrossRef]
- Hebach, C.; Kazmaier, U. Via Ugi reactions to conformationally fixed cyclic peptides. Chem. Commun. 2003, 596–597. [Google Scholar] [CrossRef]
- Marquarding, D.; Hoffmann, P.; Heitzer, H.; Ugi, I. Stereoselective syntheses. V. Isonitriles. XXIV. Stereoselective four-component condensations of a-ferrocenylethylamine and its absolute configuration. J. Am. Chem. Soc. 1970, 92, 1969–1971. [Google Scholar] [CrossRef]
- Urban, R.; Ugi, I. Asymmetrically induced four component condensation with extremely high stereoselectivity and multiplication of stereoselectivity. Angew. Chem. Int. Ed. Engl. 1975, 14, 61–62. [Google Scholar] [CrossRef]
- Herrmann, R.; Hübener, G.; Siglmüller, F.; Ugi, I. Chirale a-ferrocenylalkylamine. Liebigs Ann. Chem. 1986, 251–268. [Google Scholar]
- Siglmüller, F.; Herrmann, R.; Ugi, I. Chiral ferrocenylalkylamines from (−)-menthone. Tetrahedron 1986, 42, 5931–5940. [Google Scholar] [CrossRef]
- Kunz, H.; Pfrengle, W. Asymmetric synthesis on carbohydrate templates: Stereoselective Ugi-synthesis of a-amino acid derivatives. J. Am. Chem. Soc. 1988, 110, 651–652. [Google Scholar] [CrossRef]
- Kunz, H.; Pfrengle, W. Carbohydrates as chiral templates: Asymmetric Ugi-synthesis of alpha-amino acids using galactosylamines as the chiral matrices. Tetrahedron 1988, 44, 5487–5494. [Google Scholar] [CrossRef]
- Szardenings, A.K.; Burkoth, T.S.; Lu, H.H.; Tien, D.W.; Campbell, D.A. A simple procedure for the solid phase synthesis of diketopiperazine and diketomorpholine derivatives. Tetrahedron 1997, 53, 6573–6593. [Google Scholar] [CrossRef]
- Yamada, T.; Motoyama, N.; Taniguchi, T.; Kazuta, Y.; Miyazawa, T.; Kuwata, S.; Matsumoto, K.; Sugiura, M. Synthesis of peptides containing a,a'-iminodicarboxylic acids. Chem. Lett. 1987, 723–726. [Google Scholar]
- Faggi, C.; Marcaccini, S.; Pepino, R.; Cruz Pozo, M. Studies on isocyanides and related compounds; Synthesis of 1,4-benzodiazepine-2,5-diones via Ugi four-component condensation. Synthesis 2002, 2756–2760. [Google Scholar]
- Demharter, A.; Hörl, W.; Herdtweck, E.; Ugi, I. Synthesis of chiral 1,1'-iminodicarboxylic acid derivatives from a-amino acids, aldehydes, isocyanides, and alcohols by the diastereoselective five-center-four-component reaction. Angew. Chem. Int. Ed. Engl. 1996, 35, 173–175. [Google Scholar] [CrossRef]
- Dyker, G. Amino acid derivatives by multicomponent reactions. Angew. Chem. Int. Ed. Engl. 1997, 36, 1700–1702. [Google Scholar] [CrossRef]
- Dyker, G.; Breitenstein, K.; Henkel, G. Synthesis of novel chiral ligands from amino acids by the Ugi reaction. Tetrahedron: Asymmetry 2002, 13, 1929–1936. [Google Scholar] [CrossRef]
- Park, S.J.; Keum, G.; Kang, S.B.; Koh, H.Y.; Kim, Y.; Lee, D.H. A facile synthesis of N-carbamoylmethyl-a-aminobutyrolactones by the Ugi multicomponent condensation reaction. Tetrahedron Lett. 1998, 39, 7109–7112. [Google Scholar] [CrossRef]
- Godet, T.; Bonvin, Y.; Vincent, G.; Merle, D.; Thozet, A.; Ciufolini, M.A. Titanium catalysis in the Ugi reaction of a-amino acids with aromatic aldehydes. Org. Lett. 2004, 6, 3281–3284. [Google Scholar] [CrossRef]
- Basso, A.; Banfi, L.; Riva, R. A marriage of convenience: Combining the power of isocyanide-based multicomponent reactions with the versatility of (hetero)norbornene chemistry. Eur. J. Org. Chem. 2010, 1831–1841. [Google Scholar]
- Ziegler, T.; Kaisers, H.-J.; Schlömer, R.; Koch, C. Passerini and Ugi reactions of benzyl and acetyl protected isocyanoglucoses. Tetrahedron 1999, 55, 8397–8408. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Zhdanko, A.G.; Orru, R.V.A.; Nenajdenko, V.G. Isocyanoacetate derivatives: synthesis, reactivity, and application. Chem. Rev. 2010, 110, 5235–5331. [Google Scholar] [CrossRef]
- Floyd, C.D.; Harnett, L.A.; Miller, A.; Patel, S.; Saroglou, L.; Whittaker, M. Rapid synthesis of matrix metalloproteinase inhibitors via Ugi four-component condensation. Synlett 1998, 637–639. [Google Scholar]
- Golebiowski, A.; Klopfenstein, S.R.; Shao, X.; Chen, J.J.; Colson, A.-O.; Grieb, A.L.; Russell, A.F. Solid-supported synthesis of a peptide b-turn mimetic. Org. Lett. 2000, 2, 2615–2617. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Reznichenko, A.L.; Balenkova, E.S. Diastereoselective Ugi reaction without chiral amines: the synthesis of chiral pyrroloketopiperazines. Tetrahedron 2007, 63, 3031–3041. [Google Scholar] [CrossRef]
- France, S.; Guerin, D.J.; Miller, S.J.; Lectka, T. Nucleophilic chiral amines as catalysts in asymmetric synthesis. Chem. Rev. 2003, 103, 2985–3012. [Google Scholar] [CrossRef]
- Wurz, R.P. Chiral dialkylaminopyridine catalysts in asymmetric synthesis. Chem. Rev. 2007, 107, 5570–5595. [Google Scholar] [CrossRef]
- Vedejs, E.; Jure, M. Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed. 2005, 44, 3974–4001. [Google Scholar] [CrossRef]
- Chen, Y.; McDaid, P.; Deng, L. Asymmetric alcoholysis of cyclic anhydrides. Chem. Rev. 2003, 103, 2965–2984. [Google Scholar] [CrossRef]
- Alba, A.-N.R.; Rios, R. Oxazolones in organocatalysis, new tricks for an old reagent. Chem. Asian J. 2011, 6, 720–734. [Google Scholar] [CrossRef]
- Kagan, H.B.; Fiaud, J.C. Kinetic Resolution. In Topics in Stereochemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1988; pp. 249–330. [Google Scholar]
- Díez, D.; Gil, M.J.; Moro, R.F.; Garrido, N.M.; Marcos, I.S.; Basabe, P.; Sanz, F.; Broughton, H.B.; Urones, J.G. Chemistry of sulfones: Synthesis of a new chiral nucleophilic catalyst. Tetrahedron: Asymmetry 2005, 16, 2980–2985. [Google Scholar] [CrossRef]
- Pflugrath, J.W. CrystalClear: Rigaku Corporation, 1999. CrystalClear Software User's Guide, Molecular Structure Corporation, 2000. Acta Cryst. 1999, D55, 1718–1725. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; De Caro, L.; Giacovazzo, C.; Polidori, G.; Spagna, R. SIR2004: An improved tool for crystal structure determination and refinement. J. Appl. Cryst. 2005, 38, 381–388. [Google Scholar] [CrossRef]
- Least Squares function minimized: (SHELXL97) Sw(Fo2 − Fc2)2 where w = Least Squares weights.
- Larson, A.C. Crystallographic Computing; Ahmed, F.R., Ed.; Munksgaard: Copenhagen, Denmark, 1970; pp. 291–294, (equation 22, with V replaced by the cell volume). [Google Scholar]
- Standard deviation of an observation of unit weight: [Sw(Fo2 − Fc2)2/(No-Nv)]1/2. where: No = number of observations Nv = number of variables.
- Flack, H.D. On enantiomorph-polarity estimation. Acta Cryst. 1983, A39, 876–881. [Google Scholar]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; The Kynoch Press: Birmingham, UK, 1974; Volume IV, Table 2.2 A. [Google Scholar]
- Ibers, J.A.; Hamilton, W.C. Dispersion corrections and crystal structure refinements. Acta Cryst. 1964, 17, 781–782. [Google Scholar] [CrossRef]
- Creagh, D.C.; McAuley, W.J. International Tables for Crystallography; Wilson, A.J.C., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1992; Volume C, pp. 219–222, Table 4.2.6.8. [Google Scholar]
- Creagh, D.C.; Hubbell, J.H. International Tables for Crystallography; Wilson, A.J.C., Ed.; Kluwer Academic Publishers: Boston, MA, USA, 1992; Volume C, pp. 200–206, Table 4.2.4.3. [Google Scholar]
- Kabuto, C.; Akine, S.; Nemoto, T.; Kwon, E. Release of software (Yadokari-XG 2009) for crystal structure analyses. J. Cryst. Soc. Jpn. 2009, 51, 218–224. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mandai, H.; Irie, S.; Mitsudo, K.; Suga, S. Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions. Molecules 2011, 16, 8815-8832. https://doi.org/10.3390/molecules16108815
Mandai H, Irie S, Mitsudo K, Suga S. Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions. Molecules. 2011; 16(10):8815-8832. https://doi.org/10.3390/molecules16108815
Chicago/Turabian StyleMandai, Hiroki, Shunsuke Irie, Koichi Mitsudo, and Seiji Suga. 2011. "Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions" Molecules 16, no. 10: 8815-8832. https://doi.org/10.3390/molecules16108815
APA StyleMandai, H., Irie, S., Mitsudo, K., & Suga, S. (2011). Studies on the Synthesis of DMAP Derivatives by Diastereoselective Ugi Reactions. Molecules, 16(10), 8815-8832. https://doi.org/10.3390/molecules16108815