1. Introduction
Heterocycles are important structural units found in a wide range of biologically active compounds [
1]. There have been many calls for the synthesis of novel heterocyclic systems to be used as building blocks for the next generation of pharmaceuticals [
1,
2,
3,
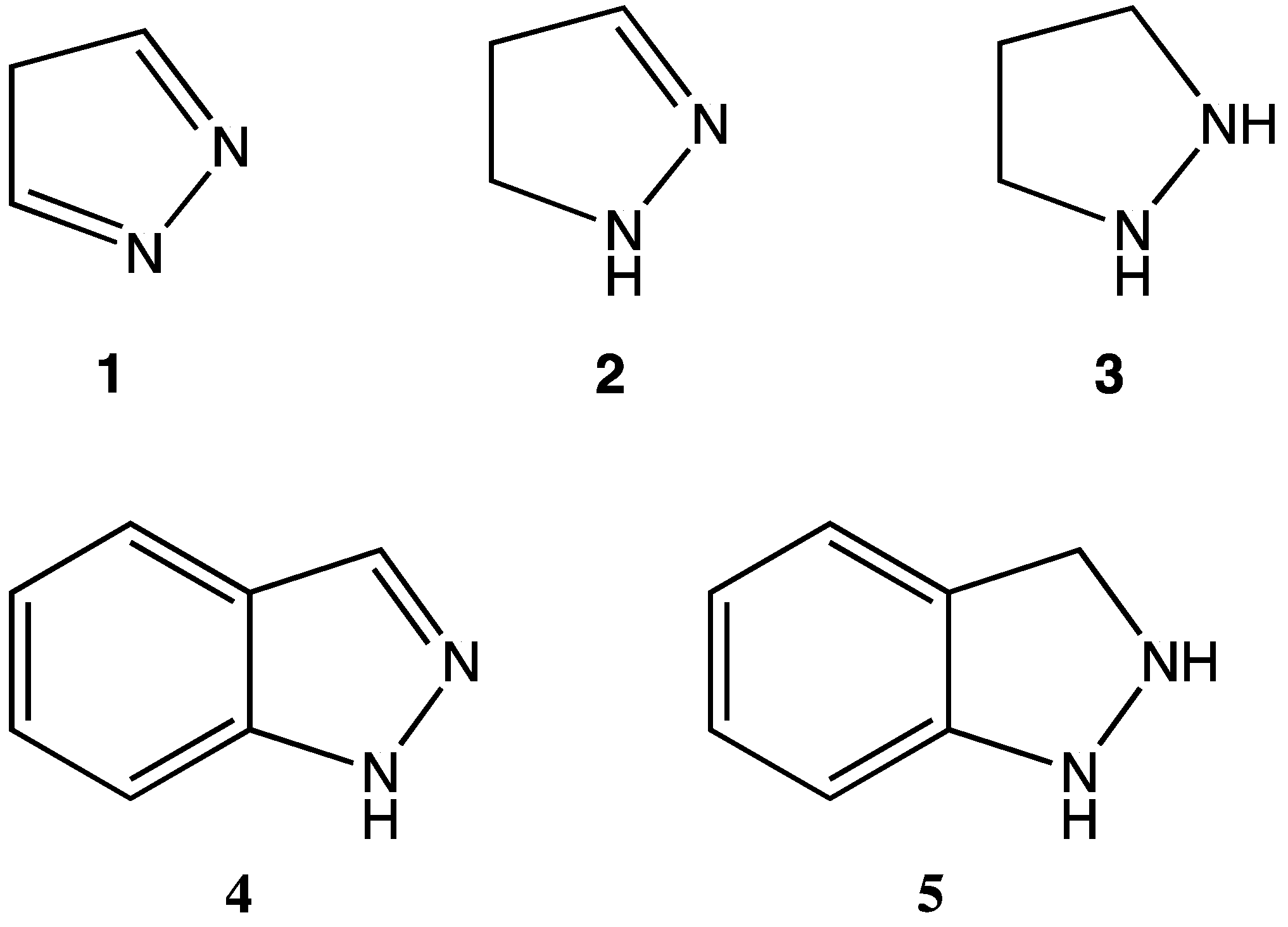
4]. One subclass of particularly active heterocycles are those bearing 1,2-dinitrogen substitution. For example, substituted derivatives of the pyrazole (
1 in
Figure 1), pyrazoline (
2), and pyrazolidine (
3) ring systems are of pharmaceutical interest for their demonstrated antibacterial, antidepressant, and/or anti-inflammatory activities (among others) [
4,
5,
6]. Additionally, the fused aromatic 1
H-indazole (
4) is well recognized for its antihypertensive and anticancer properties [
7]. Surprisingly, however, the 2,3-dihydro-1
H-indazole moiety (
5 in
Figure 1) remains virtually unknown and untested, despite its obvious structural similarities to both the pyrazolidine (
3) and 1
H-indazole (
4) ring systems.
Figure 1.
Structures of select 1,2-dinitrogen heterocycles.
Figure 1.
Structures of select 1,2-dinitrogen heterocycles.
We are aware of only a handful of studies that have addressed the synthesis of derivatives of indazoles
5 [
8,
9,
10,
11,
12,
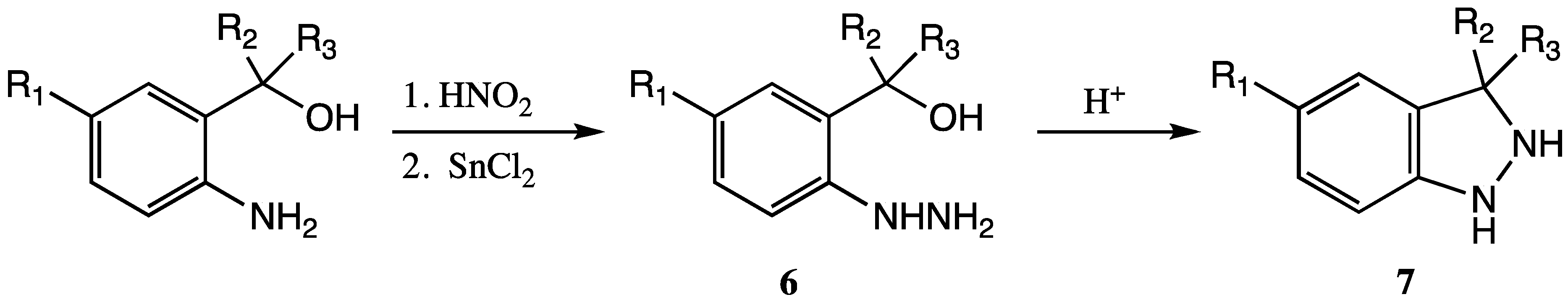
13]. Zenchoff reported on their synthesis via acid-catalyzed S
N1-type ring closure of substituted 2-hydroxymethyl phenylhydrazines
6 to form indazoles
7 (
Scheme 1) [
8]. Overall yields of approximately 30% were reported for the combined two-step method.
Scheme 1.
Synthesis of substituted derivatives of 2,3-dihydro-1H-indazoles 7 via acid-catalyzed ring-closure.
Scheme 1.
Synthesis of substituted derivatives of 2,3-dihydro-1H-indazoles 7 via acid-catalyzed ring-closure.
Additionally, there have been a few studies in which formation of derivatives of indazole
5 have been observed as a result of trapping experiments on reactive intermediates [
9,
10,
11]. However, other than Zenchoff’s limited work, we are unaware of the development of any general routes for the synthesis of this class of heterocycles.
In addition to being interesting compounds in their own right, 2,3-dihydro-1
H-indazoles
5 have also served as synthetic intermediates for their more thoroughly-studied counterparts, the 1
H-indazoles
4 [
8]. Thus, development of synthetic routes towards the synthesis of indazoles
5 similarly creates novel routes for the synthesis of indazoles
4.
The sparse number of investigations into the synthesis of this class of heterocycles is, therefore, quite surprising given that 2,3-dihydro-1H-indazoles have such strong potential for biological activity and as convenient synthetic intermediates. Detailed investigations of their properties, however, demand robust synthetic methods for their preparation. Herein, we describe our initial investigations towards the synthesis of this understudied class of heterocyclic compounds.
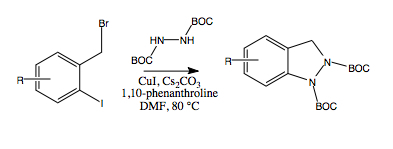
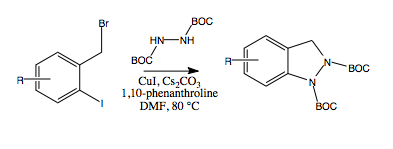
2. Results and Discussion
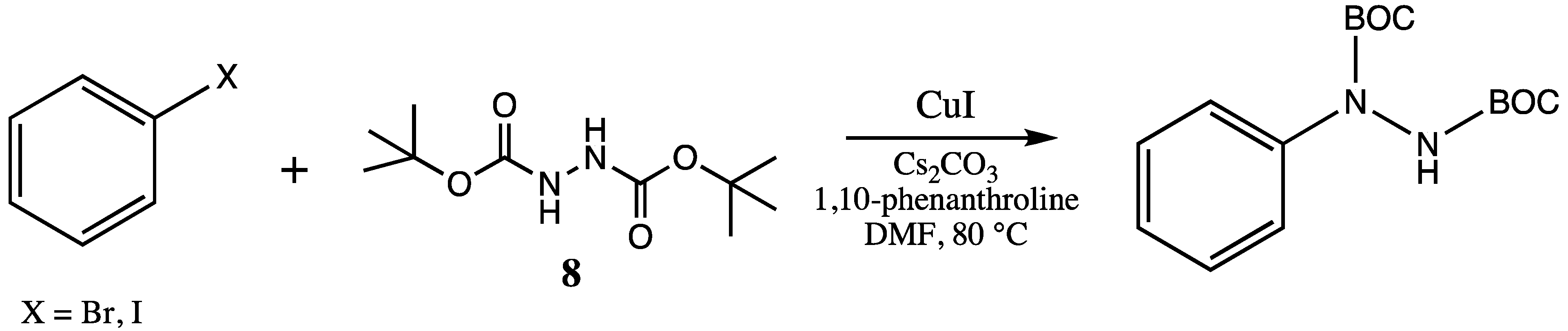
Ullman-type copper(I)-mediated coupling of bis-BOC protected hydrazine
8 to aryl and vinyl halides in DMF using CuI, 1,10-phenanthroline and Cs
2CO
3 is a well-established synthetic procedure (see
Scheme 2) [
14,
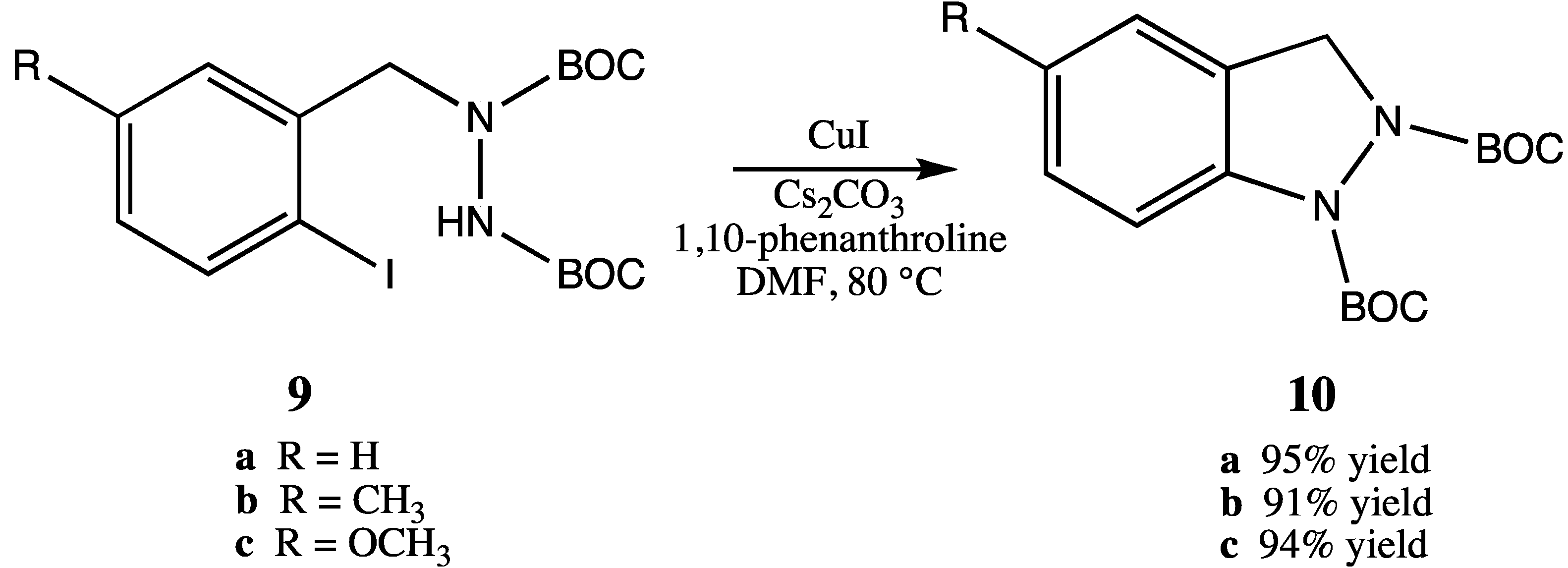
15]. We initially envisioned the synthesis of bis-BOC protected 2,3-dihydro-1
H-indazoles
10 via intramolecular coupling of the free N-H bond of hydrazines
9 at the iodo-substituted carbon atom to form the five-membered indazole nucleus (
Scheme 3). Indeed, this proved to be an excellent method for the synthesis of substituted indazoles, providing high yields of
10 (
Scheme 3).
Scheme 2.
Ullman-type coupling of hydrazine 8 to halogenated aromatics.
Scheme 2.
Ullman-type coupling of hydrazine 8 to halogenated aromatics.
Scheme 3.
Intramolecular coupling of hydrazines 9 to form bis-BOC protected indazoles 10.
Scheme 3.
Intramolecular coupling of hydrazines 9 to form bis-BOC protected indazoles 10.
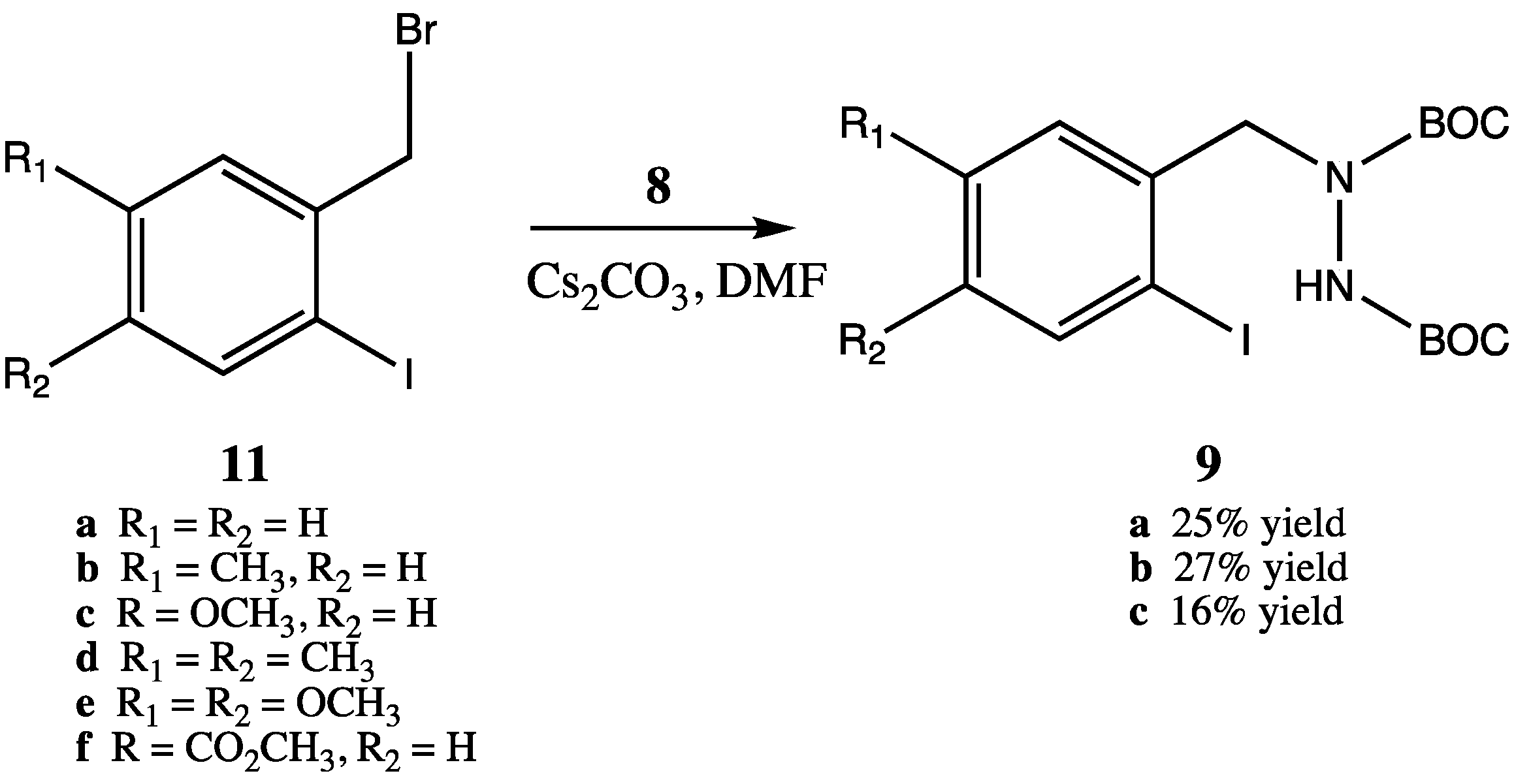
Unfortunately, however, synthesis of the required trisubstituted hydrazine starting materials
9 proved to be problematic (
Scheme 4). Although we utilized reaction conditions optimized by Rasmussen for selective monoalkylation of
8 [
16], only poor yields of monoalkylated product were obtained (
i.e., compounds
10a–c were obtained in 25%, 27% and 16% yields, respectively). Surprisingly, bisalkylation of hydrazine
8 was the preferred reaction route. We attempted to increase the yield of monoalkylated product by increasing the amount of starting
8 relative to starting benzyl bromides
11, but this led to difficulties in separation of the product from excess
8. Additionally, other than
11a, which was commercially available, the required substituted
ortho-iodobenzyl bromides (compounds
11b–f) needed to be synthesized from the corresponding benzylic alcohols via a two step process of iodination of the aromatic ring (silver CF
3CO
2Ag/I
2) followed by bromination at the benzylic alcohol position (PBr
3). Thus, although the intramolecular coupling reactions afforded high yields (
Scheme 3), the effective yields from the synthetically expensive ortho-iodobenzyl bromides were unsatisfactorily low.
Scheme 4.
Synthesis of monoalkylated bis-BOC protected hydrazines 9.
Scheme 4.
Synthesis of monoalkylated bis-BOC protected hydrazines 9.
To bypass the problem of dialkylation, we considered the possibility of developing a one-pot procedure in which copper-mediated coupling of the initially formed monoalkylated hydrazines 9 to form indazoles 10 might be able to compete with the complicating dialkylation process. A mixture of 8 and ortho-iodobenzyl bromide 11a was added slowly, via syringe pump (over a period of 7 h), to a pre-heated stirring mixture of CuI, 1,10-phenanthroline and Cs2CO3. After 24 h, we were gratified to note that the only major product detected by TLC analysis of the crude reaction mixture was the desired indazole 10a. Reaction workup, followed by column chromatography afforded 10a in 60% yield, a substantial increase from the effective 22% yield obtained via the two-step process (i.e., 11a→9a→10a) starting from the ortho-iodobenzyl bromide 11a. In addition, this procedure avoided the need for purification of the intermediate hydrazine 9a. Similarly, indazoles 9b and 9c were formed in 60% yields utilizing the one-pot procedure rather than the effective 25% and 15% yields, respectively, from the corresponding two-step procedures.
Given the success of this one-pot procedure, we subjected a number of substituted ortho-iodobenzyl bromides to the same reaction conditions. The yields were consistent at ~60% (
Table 1), and the products were obtained in pure form following column chromatography. The reaction was tolerant of electron-donating (
i.e., OCH
3) and electron-withdrawing (
i.e., CO
2CH
3) groups.
Table 1.
One-pot synthesis of substituted bis-BOC protected indazoles
10 starting from
ortho-iodo benzylbromides
11.
![Molecules 16 09553 i001]()
Table 1.
One-pot synthesis of substituted bis-BOC protected indazoles 10 starting from ortho-iodo benzylbromides 11. ![Molecules 16 09553 i001]()
| Entry | Substrate | Indazole Product | Yield (%) |
|---|
| 1 | 11a | 10a | 60 |
| 2 | 11b | 10b | 60 |
| 3 | 11c | 10c | 60 |
| 4 | 11d | 10d | 55 |
| 5 | 11e | 10e | 72 |
| 6 | 11f | 10f | 62 |
3. Experimental
3.1. General
All chemicals and solvents were used as received (Aldrich, St Louis, MO, USA) Anhydrous DMF was kept under nitrogen and sealed with a septum. Column chromatography was performed using 230–400 mesh silica gel 60. NMR spectra were recorded on a Varian 60 MHz instrument in CDCl
3 as solvent, unless otherwise indicated, and referenced relative to TMS (0.0 PPM). Combustion analysis was performed by Micro Analysis Inc. (Wilmington, DE, USA). Other than unsubstituted 2-iodobenzyl bromide
11a, which was commercially available (Aldrich), benzyl bromides
11b–f were synthesized via standard aromatic iodination (I
2, CF
3CO
2Ag) followed by bromination of the benzylic alcohol (PBr
3) following procedures described in the literature [
17].
3.2. Synthesis of Alkylated Monoalkylated Hydrazines 9a–c
Representative Procedure: Preparation of 1,2-di-tert-Butyl 1-(2-iodobenzyl)-1,2-hydrazinedicarboxylate (9a) To a solution of di-tert-butyl hydrazodiformate (8, 0.71 g, 3.06 mmol) in anhydrous DMF (15 mL) was added Cs2CO3 (2 g, 2 equiv.) followed immediately by 2-iodobenzyl bromide (11a, 1 g, 3.37 mmol, 1.1 equiv.). The mixture was stirred for 2 h, after which time TLC indicated consumption of the bromide. The mixture was diluted with H2O (25 mL) and washed with EtOAc (3 × 25 mL). The combined EtOAc layers were backwashed with brine (3 × 25 mL), dried and concentrated. Column chromatography (SiO2, 4:1 hexanes/EtOAc as eluent) afforded 0.32 g (23% yield) of 9a as a white solid. 1H-NMR δ 7.82 (d, J = 7.4 Hz, 1H), 7.34–6.79 (m, 3H), 6.45 (br s, NH, 1H), 4.71 (s, 2H), 1.48 (s, 9H), 1.45 (s, 9H). 13C-NMR δ 155.3, 155.0, 139.5, 139.3, 129.7, 129.1, 128.3, 99.0, 81.5, 81.1, 58.1, 28.2. Anal. Calcd. for C17H25N2O4I: C, 45.53; H, 5.62; N, 6.25; H. Found: C, 45.61; H, 5.56; N, 6.12.
1,2-Di-tert-butyl 1-(2-iodo-5-methylbenzyl)-1,2-hydrazinedicarboxylate (9b). Compound 9b (0.46 g, 27% yield) was isolated as a white solid following the representative procedure described above starting with 2-iodo-5-methylbenzyl bromide (11b, 1.1 g, 3.37 mmol). 1H-NMR δ 7.64 (d, J = 8.0 Hz, 1H), 7.10 (d, J = 1.7 Hz, 1H), 6.74 (dd, J = 8.0, 1.7 Hz, 1H), 6.67 (br s, NH, 1H), 4.67 (s, 2H), 2.27 (s, 3H), 1.48 (s, 9H), 1.44 (s, 9H). 13C-NMR δ 155.1, 154.7, 138.9, 138.6, 137.8, 130.2, 129.9, 94.7, 81.1, 80.6, 57.4, 27.9, 20.7. Anal. Calcd. for C18H27N2O4I: C, 46.74; H, 5.89; N, 6.06; H. Found: C, 46.61; H, 5.84; N, 5.96.
1,2-Di-tert-butyl 1-(2-iodo-5-methoxybenzyl)-1,2-hydrazinedicarboxylate (9c). Compound 9c (0.24 g, 16% yield) was isolated as a thick colorless liquid following the representative procedure described above starting with 2-iodo-5-methoxybenzyl bromide (11c, 1.1 g, 3.42 mmol). 1H-NMR δ 7.67 (d, J = 8.7 Hz, 1H), 6.93 (d, J = 3.0 Hz, 1H), 6.55 (dd, J = 8.7, 3.0 Hz, 1H), 6.45 (br s, NH, 1H), 4.66 (s, 2H), 3.76 (s, 3H), 1.48 (s, 9H), 1.45 (s, 9H). 13C-NMR δ 160.1, 155.3, 154.9, 140.3, 140.0, 115.6, 115.1, 87.1, 81.6, 81.2, 58.0, 55.3, 28.2. Anal. Calcd. for C18H27N2O5I: C, 45.18; H, 5.86; N, 5.69; H. Found: C, 43.81; H, 5.51; N, 5.55.
3.3. Synthesis of 1,2-Di-tert-butyl 1H-indazole-1,2-(3H)-dicarboxylates 10a–c via Intramolecular Cyclization
Representative Procedure:
Preparation of 1,2-di-tert-Butyl 5-methyl-1H-indazole-1,2-(3H)-di-carboxylate (
10b). A mixture of
9b (0.31 g, 6.7 mmol), CuI (0.13 g, 1 equiv.), 1,10-phenanthroline (0.12 g, 1 equiv.) and Cs
2CO
3 (0.33 g, 1.5 equiv.) in anhydrous DMF (5 mL) was stirred under an atmosphere of N
2 at 80 °C for 24 h. The solution was cooled, and filtered through a short column of Celite which was rinsed thoroughly with EtOAc (100 mL). The resulting mixture was filtered to remove an insoluble precipitate, concentrated to a thick brown oil, and subjected to column chromatography (SiO
2 using 4:1 hexanes/EtOAc as eluent) to afford
10b (0.20 g, 91% yield) as a thick colorless oil.
1H-NMR δ 7.42 (d,
J = 8.8 Hz, 1H), 7.10–6.99 (m, 2H), 5.18–4.45 (m, 2H), 2.32 (s, 3H), 1.56 (s, 9H), 1.50 (s, 9H).
13C-NMR δ 156.4, 153.0, 138.0, 134.0, 128.3, 122.6, 115.9, 82.2, 81.9, 51.5, 28.2, 20.9. The glassy nature of this product prevented us from obtaining a satisfactory C,H,N analysis. Evidence of purity is provided by the NMR spectra given in the
Supplementary Material.
1,2-Di-tert-butyl 1H-indazole-1,2-(3H)-dicarboxylate (10a). Compound 10a (0.21 g, 95% yield) was isolated as a white solid following the representative procedure starting with 9a (0.31 g, 0.68 mmol). 1H-NMR δ 7.64–7.07 (m, 4H), 5.22–4.51 (m, 2H), 1.57 (s, 9H), 1.51 (s, 9H). 13C-NMR δ 156.4, 152.8, 140.2, 128.2, 127.8, 124.2, 122.0, 116.1, 82.4, 82.0, 51.6, 28.2. Anal. Calcd. for C17H24N2O4: C, 63.72; H, 7.55; N, 8.75; H. Found: C, 63.81; H, 7.56; N, 8.56.
1,2-Di-tert-butyl 5-methoxy-1H-indazole-1,2-(3H)-dicarboxylate (10c). Compound 10c (0.11 g, 94% yield) was isolated as a white solid following the representative procedure starting with 9c (0.16 g, 0.34 mmol). 1H-NMR δ 7.51–7.35 (m, 1H), 6.88–6.73 (m, 2H), 5.15–4.46 (m, 2H), 3.78 (s, 3H), 1.56 (s, 9H), 1.51 (s, 9H). 13C-NMR δ 157.1, 156.3, 153.2, 133.8, 129.6, 116.9, 113.0, 108.0, 82.1, 81.8, 55.6, 51.6, 28.2. Anal. Calcd. for C18H26N2O5: C, 61.68; H, 7.48; N, 8.00; H. Found: C, 61.70; H, 7.47; N, 7.87.
3.4. Synthesis of 1,2-Di-tert-butyl 1H-indazole-1,2-(3H)-dicarboxylates 10a–f via the One-Pot Procedure
Representative Procedure: Preparation of 1,2-di-tert-Butyl 5-methoxy-1H-indazole-1,2-(3H)-di-carboxylate (10c). To a stirring mixture of CuI (0.58 g, 1 equiv.), 1,10-phenanthroline (0.55 g, 1 equiv.) and Cs2CO3 (2 g, 2 equiv.) in anhydrous DMF (10 mL) under N2 preheated to 80 °C a solution of 2-iodo-5-methoxybenzyl bromide 11c (1 g, 3.06 mmol) and di-tert-butyl hydrazodiformate (8, 1.06 g, 1.5 equiv.) in anhydrous DMF (7 mL) was added via syringe pump at a rate of 1 mL/h. The entire reaction (including injection time) was allowed to proceed for 24 h, after which time it was cooled and filtered through a short column of Celite which was rinsed thoroughly with EtOAc (150 mL). The resulting EtOAc mixture was filtered to remove an insoluble precipitate, concentrated to a thick brown oil, and subjected to column chromatography (SiO2 using 4:1 hexanes/EtOAc as eluent) to afford 0.64 g (60% yield) of 10c as a white crystalline solid.
1,2-Di-tert-butyl 1H-indazole-1,2-(3H)-dicarboxylate (10a). Compound 10a (0.97 g, 60% yield) was isolated as a white solid following the representative procedure starting with 2-iodobenzyl bromide (11a, 1.50 g, 5.05 mmol).
1,2-Di-tert-butyl 5-methyl-1H-indazole-1,2-(3H)-dicarboxylate (10b). Compound 10b (0.65 g, 60% yield) was isolated as a thick colorless liquid following the representative procedure starting with 2-iodo-5-methylbenzyl bromide (11b, 1.0 g, 3.22 mmol).
1,2-Di-tert-butyl 5,6-dimethyl-1H-indazole-1,2-(3H)-dicarboxylate (
10d). Compound
10d (0.59 g, 55% yield) was isolated as a thick colorless liquid following the representative procedure starting with 2-iodo-4,5-dimethylbenzyl bromide (11d, 1.0 g, 3.10 mmol).
1H-NMR δ 7.36 (s, 1H), 6.96 (s, 1H), 5.16–4.43 (m, 2H), 2.27 (s, 3H), 2.23 (s, 3H), 1.57 (s, 9H), 1.50 (s, 9H).
13C-NMR δ 156.5, 153.1, 138.4, 136.0, 132.4, 125.6, 122.9, 117.2, 82.0, 81.7, 51.5, 28.2, 20.1, 19.4. The glassy nature of this product prevented us from obtaining a satisfactory C,H,N analysis. Evidence of purity is provided by the NMR spectra given in the
Supplementary Material.
1,2-Di-tert-butyl 5,6-dimethoxy-1H-indazole-1,2-(3H)-dicarboxylate (
10e). Compound
10e (0.85 g, 72% yield) was isolated as a thick colorless liquid following the representative procedure starting with 2-iodo-4,5-dimethoxybenzyl bromide (
11e, 1.1 g, 3.01 mmol).
1H-NMR δ 7.36, 7.21 (s, 1H), 6.75 (s, 1H), 5.15-4.47 (m, 2H), 3.90 (s, 3H), 3.85 (s, 3H), 1.57 (s, 9H), 1.51 (s, 9H).
13C-NMR δ 156.5, 153.2, 149.0, 146.7, 134.0, 119.0, 105.9, 101.4, 82.1, 81.8, 56.5, 56.1, 51.8, 28.2. The glassy nature of this product prevented us from obtaining a satisfactory C,H,N analysis. Evidence of purity is provided by the NMR spectra given in the
Supplementary Material.
1,2-Di-tert-butyl 5-carbomethoxy-1H-indazole-1,2-(3H)-dicarboxylate (10f). Compound 10f (0.43 g, 62% yield) was isolated as a thick colorless liquid following the representative procedure starting with 2-iodo-5-carbomethoxybenzyl bromide (11f, 0.65 g, 1.83 mmol). 1H-NMR δ 8.08-7.89 (m, 2H), 7.55 (d, J = 8.3Hz, 1H), 5.02–4.75 (m, 2H), 3.90 (s, 3H), 1.58 (s, 9H), 1.51 (s, 9H). 13C-NMR δ 166.3, 156.3, 151.9, 143.8, 130.3, 128.4, 126.1, 123.7, 115.2, 83.0, 82.4, 52.0, 51.5, 28.1. Anal. Calcd. for C19H26N2O6: C, 60.29; H, 6.93; N, 7.41; H. Found: C, 60.35; H, 6.96; N, 7.15.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
