A Journey Under the Sea: The Quest for Marine Anti-Cancer Alkaloids



Abstract
:1. Introduction
2. Anti-Cancer Alkaloids Derived from Microorganisms
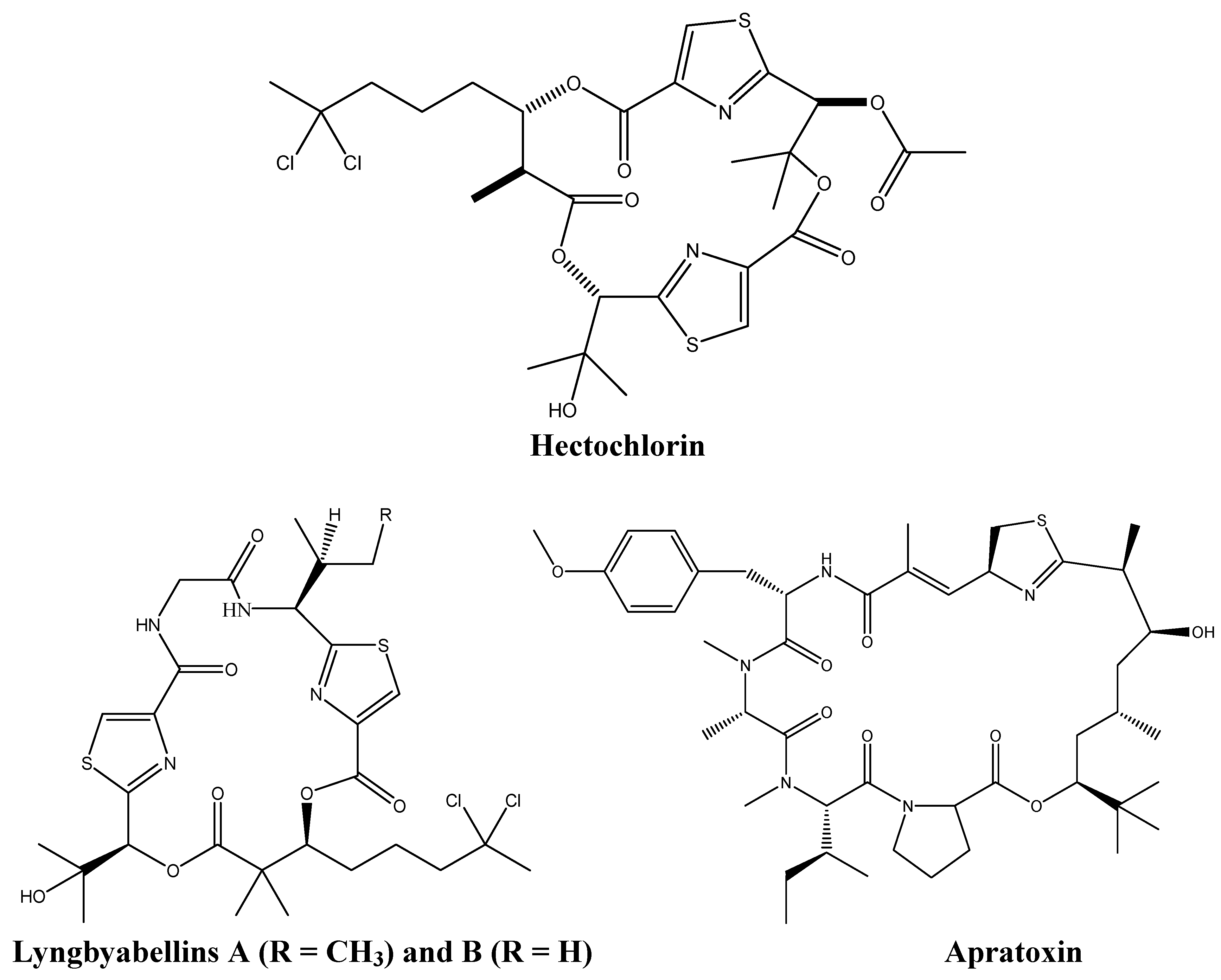
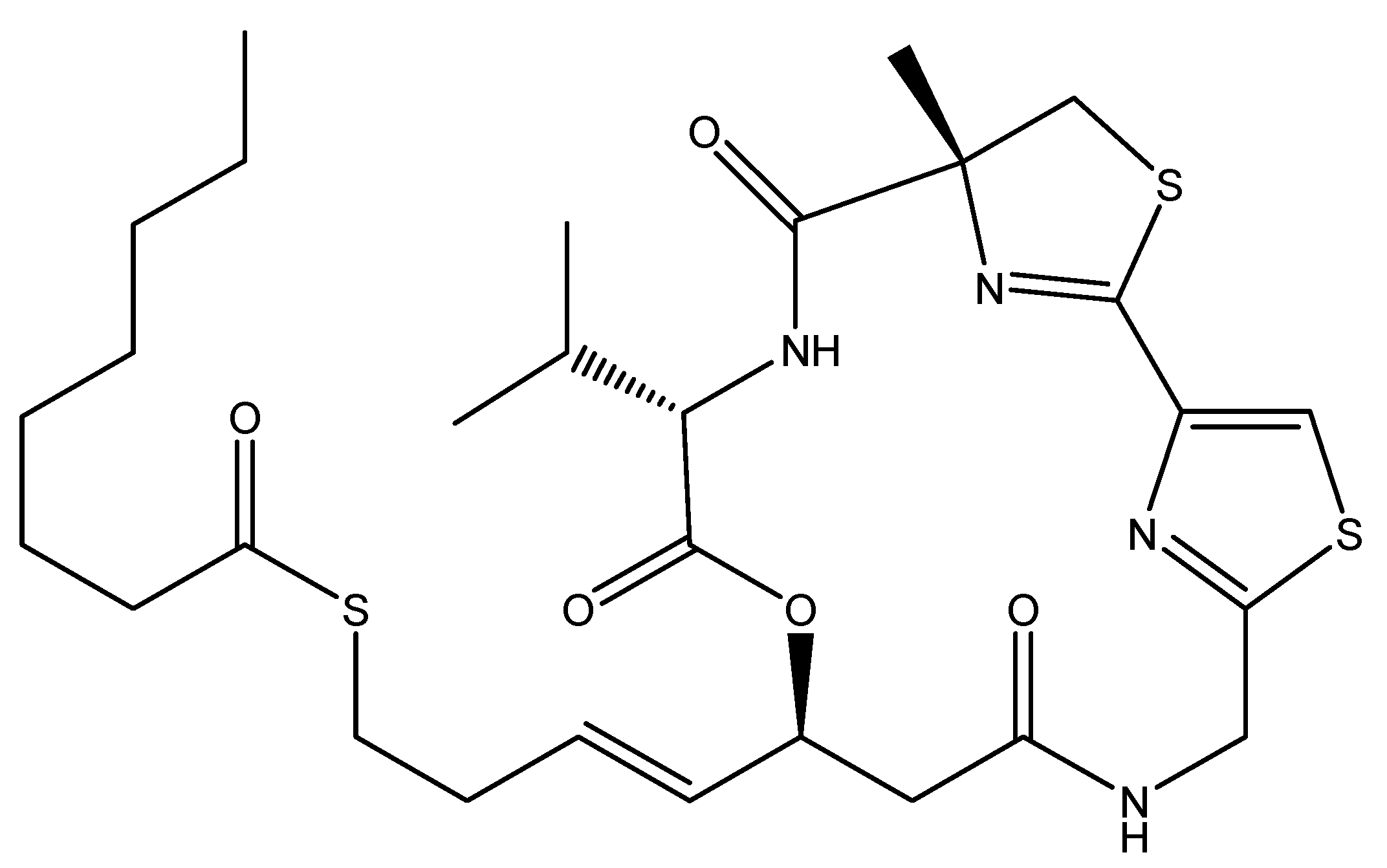
2.1. Cyanobacteria: A Pool of Bioactive Alkaloids


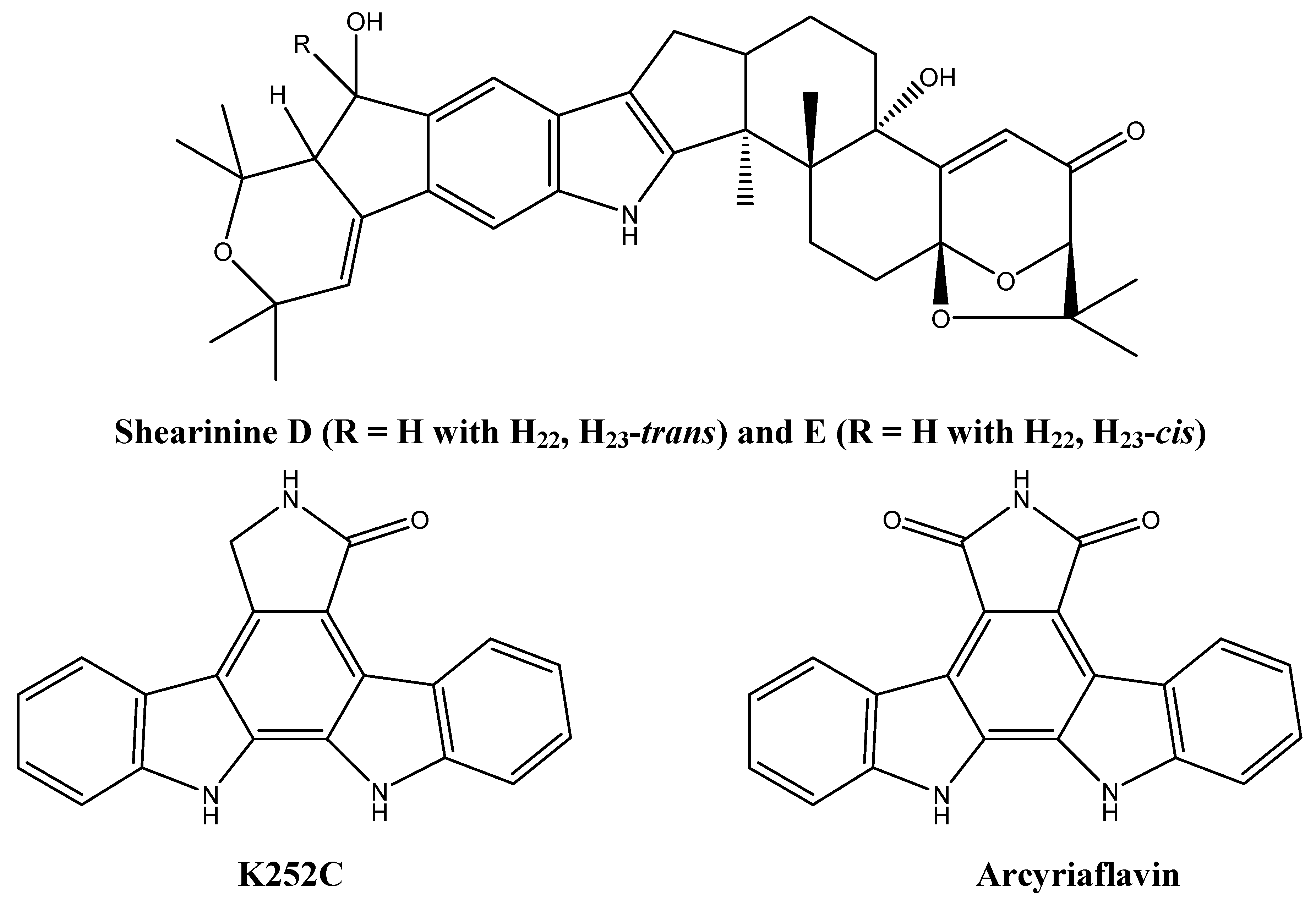
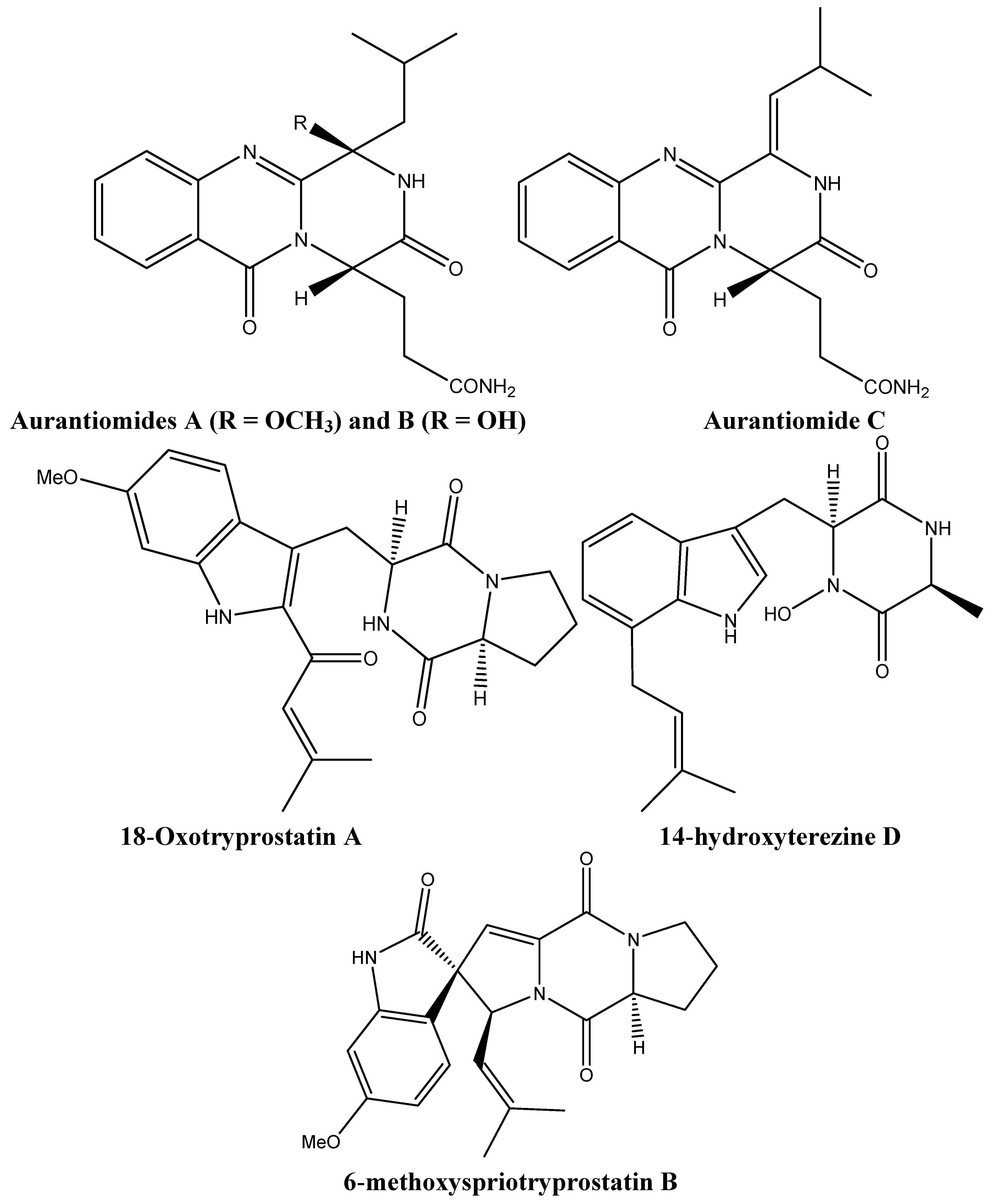
2.2. New Alkaloids from Marine Fungi: An Embryonic Stage


3. Sponge-Derived Alkaloids: Molecules with Defined Mechanisms of Action

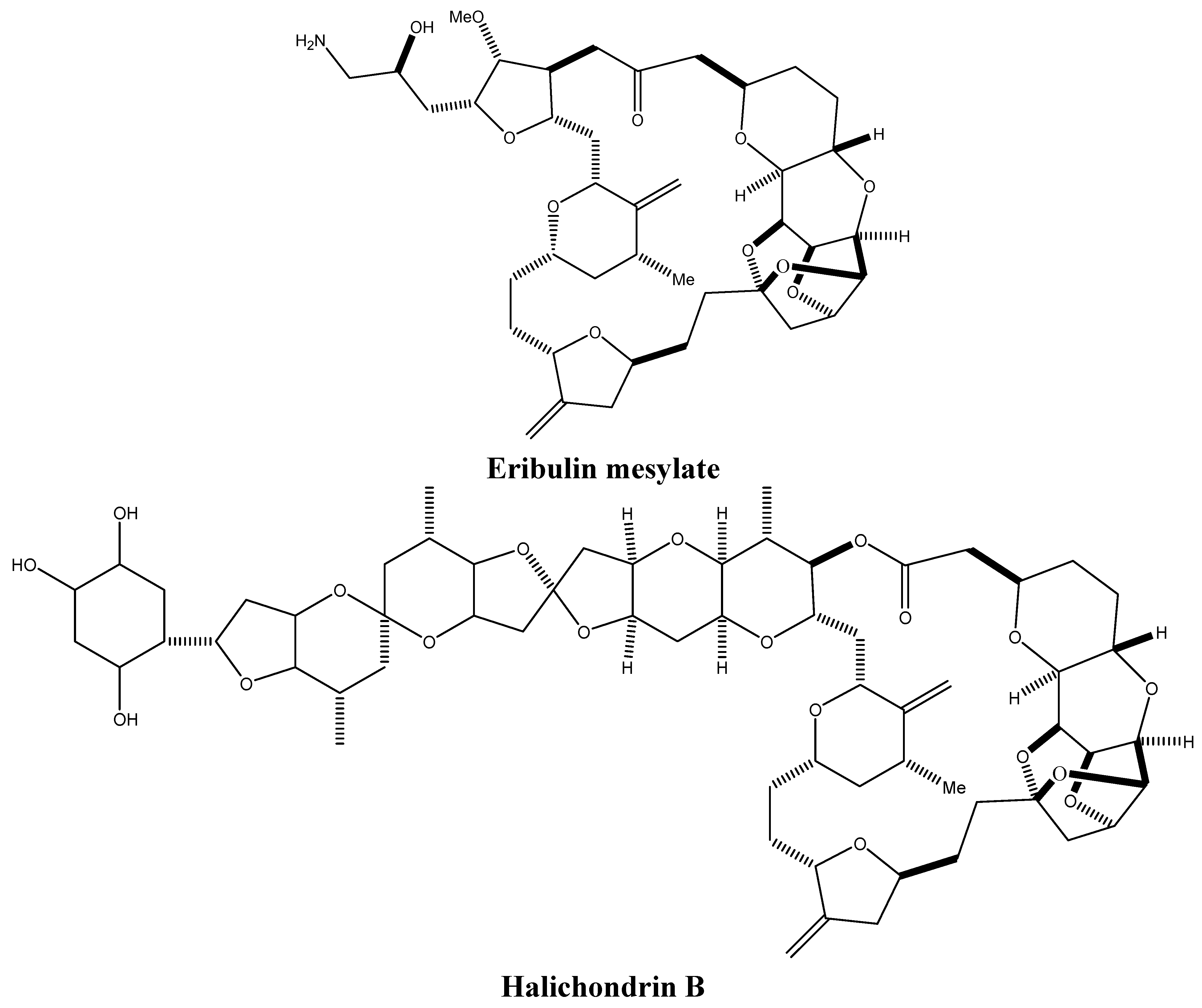
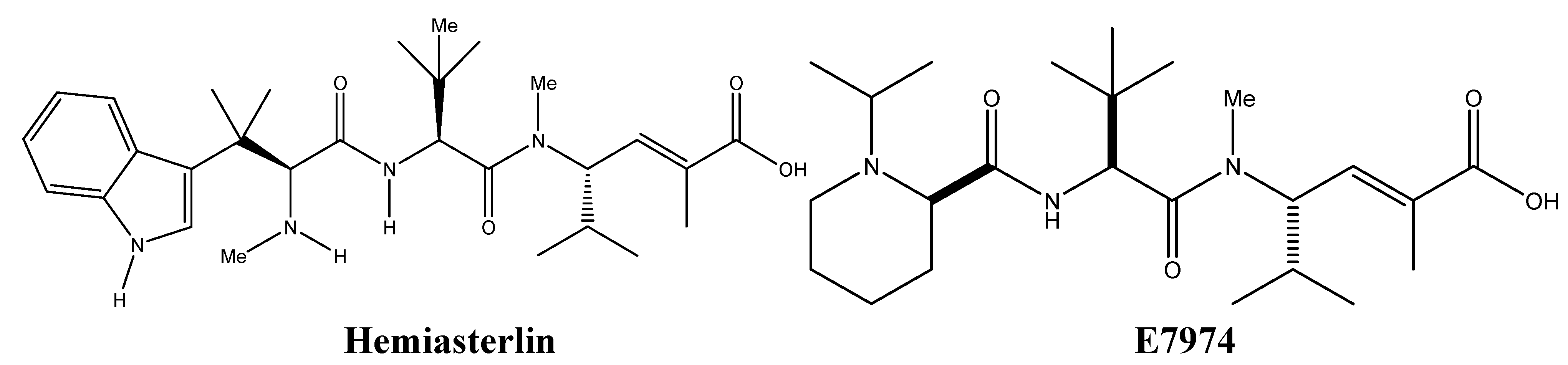
3.1. Targeting Tubulin Polymerization

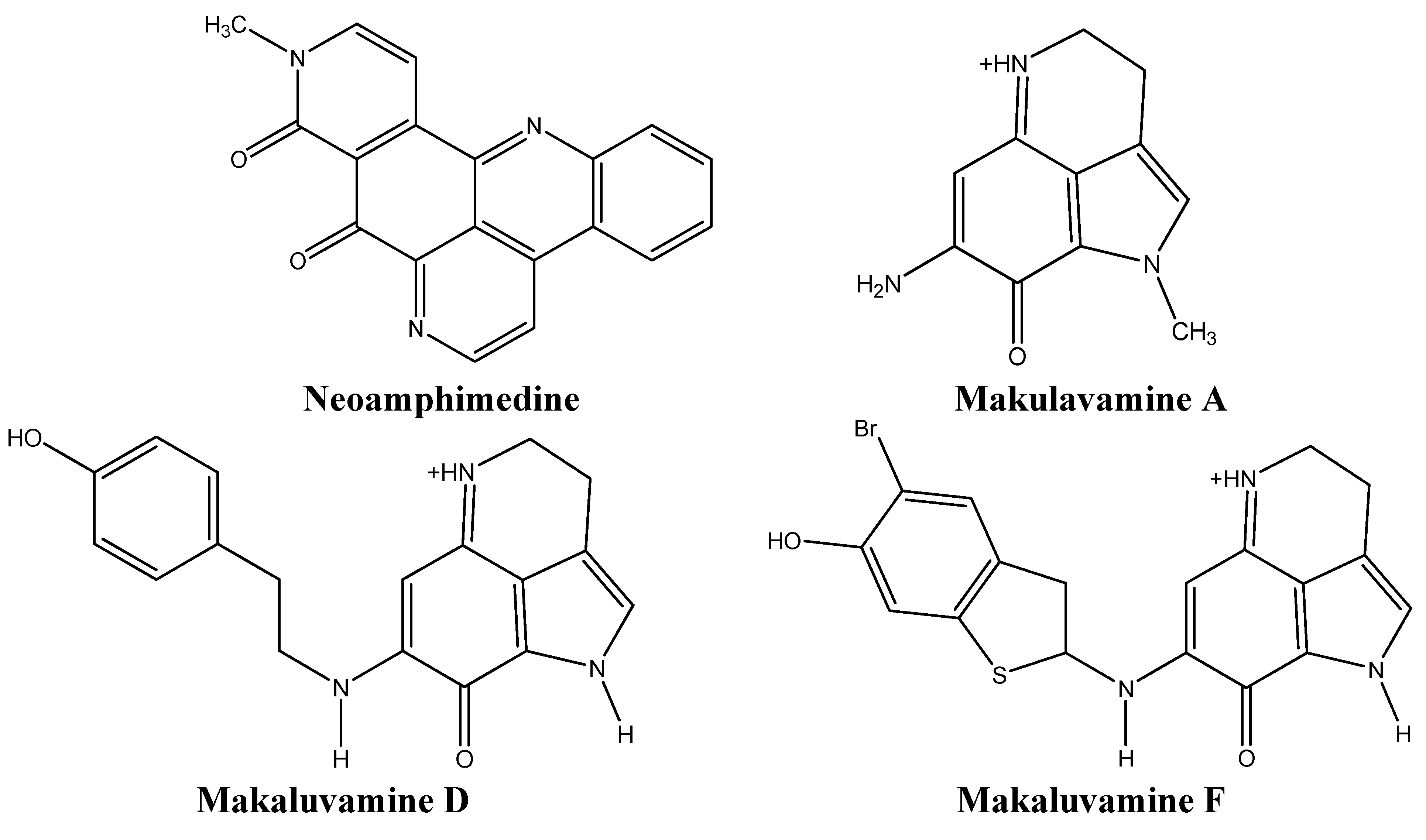
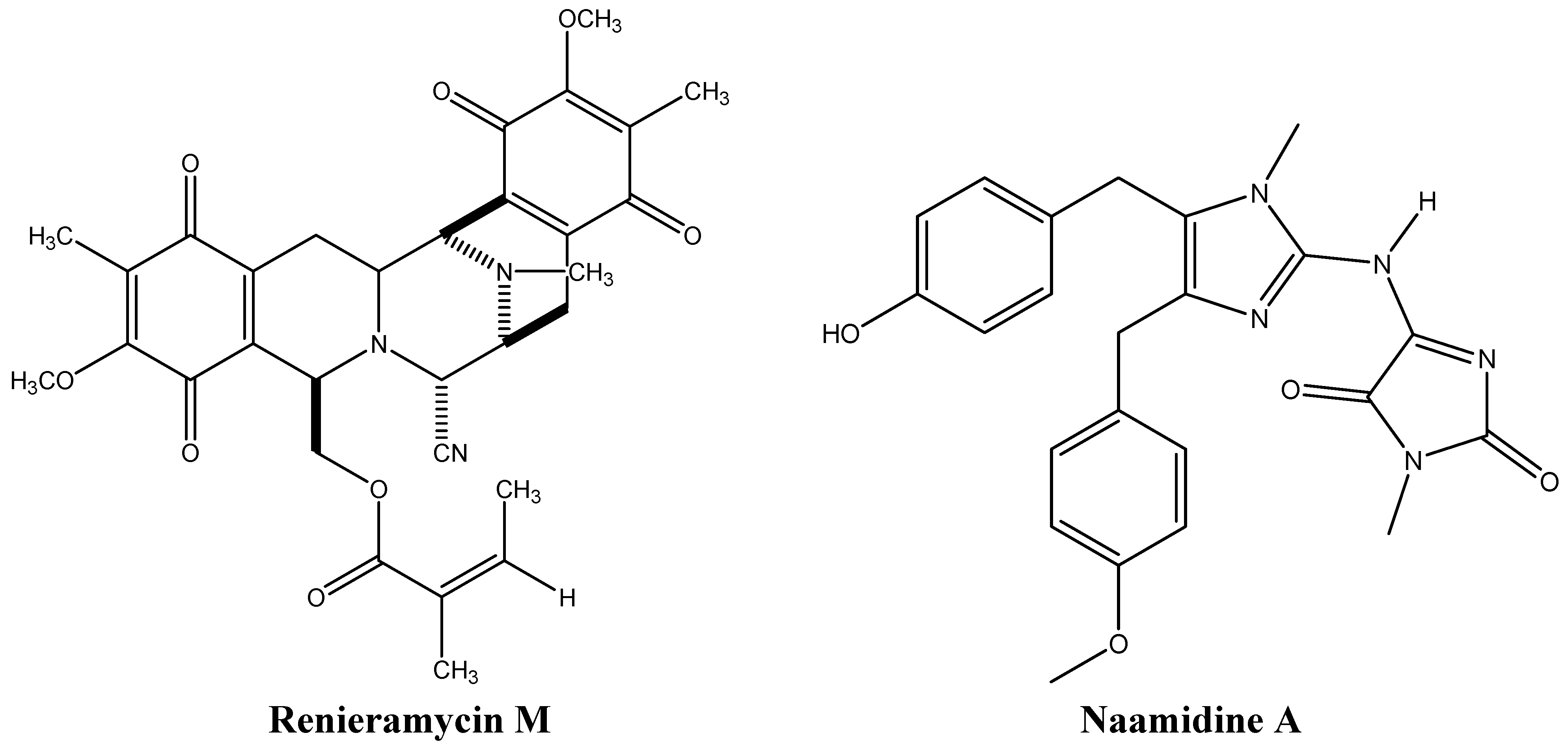
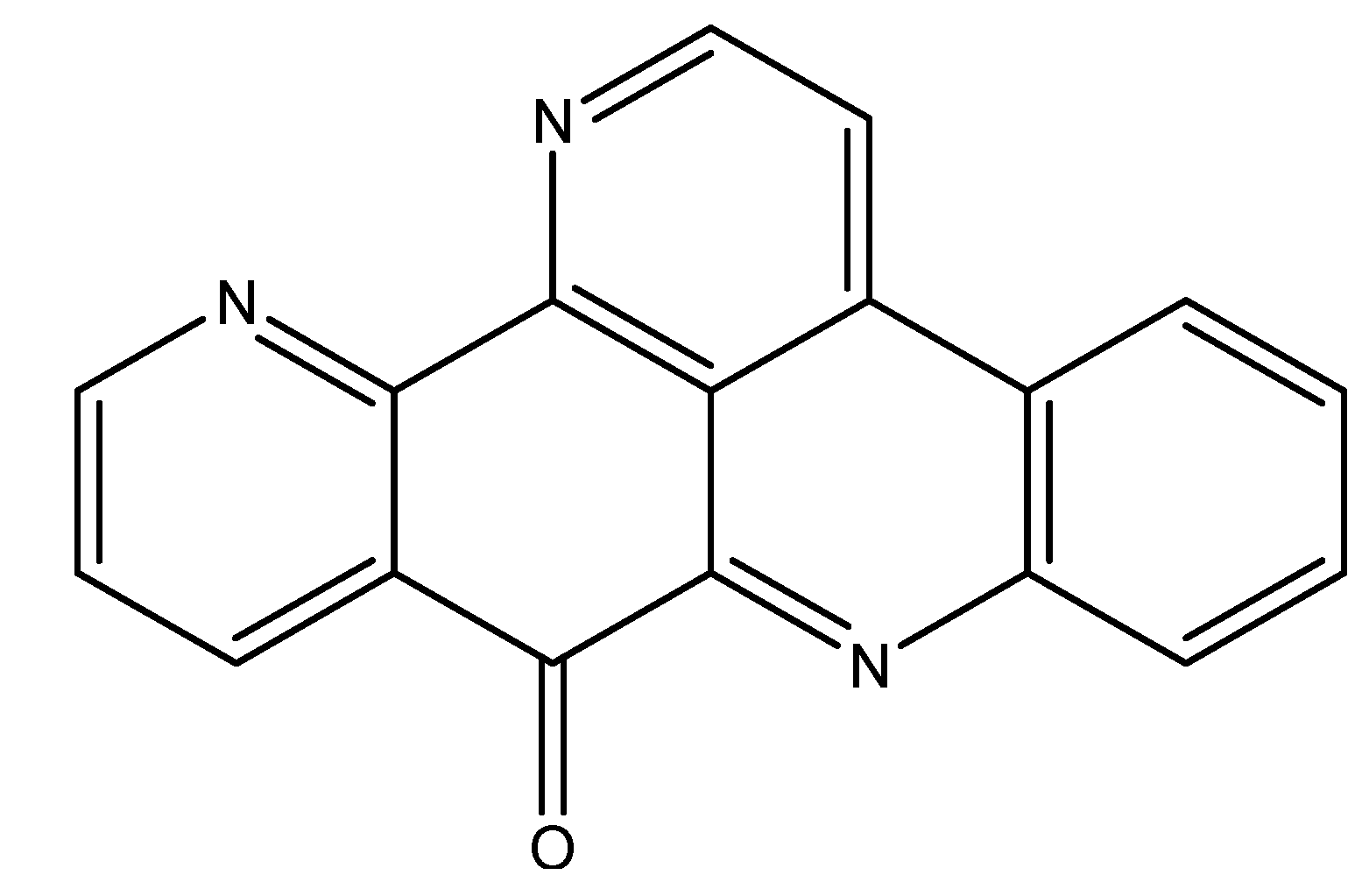
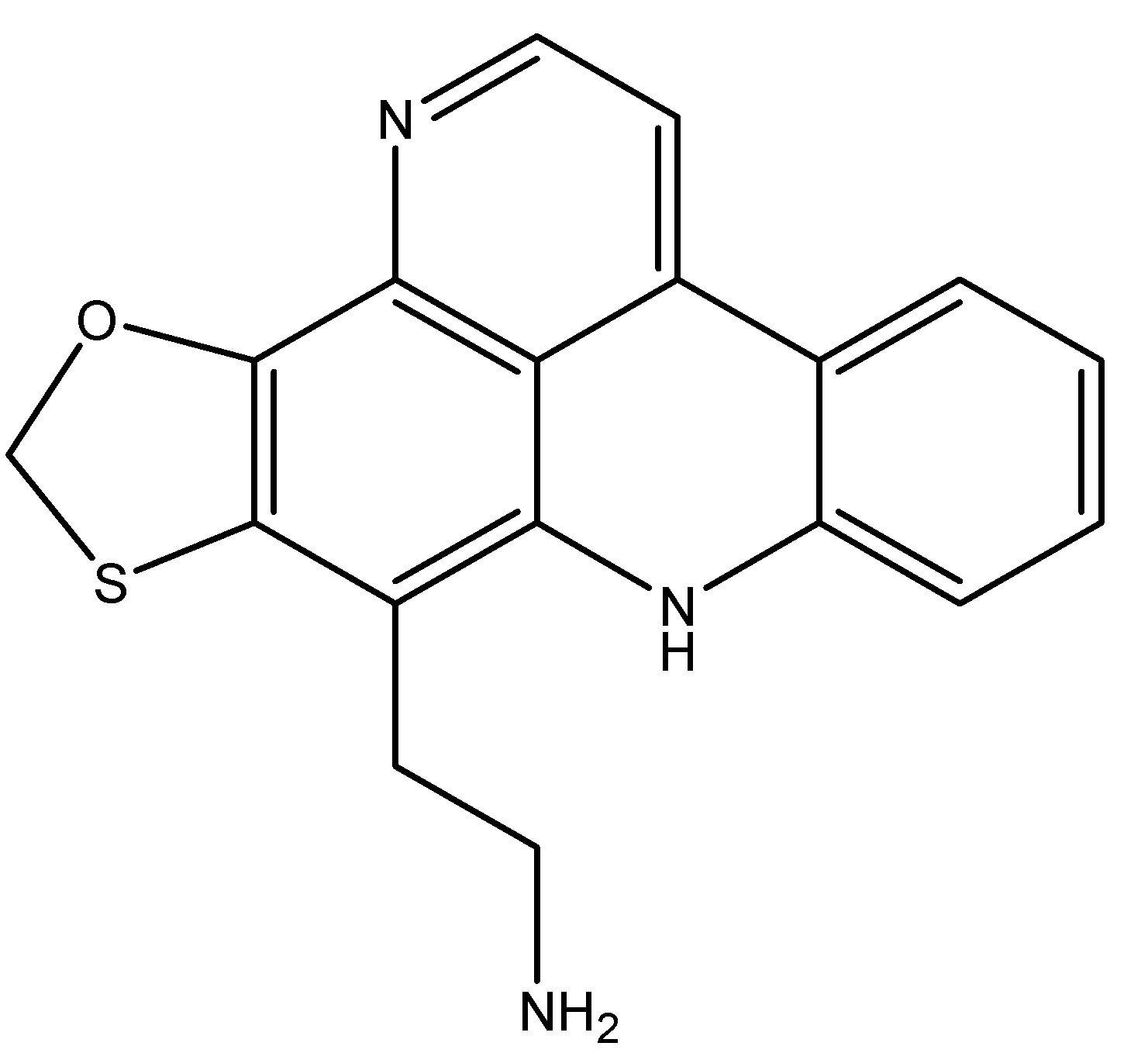
3.2. Inhibiting Topoisomerases

3.3. Targeting Molecular Players in Programmed Cell Death

3.4. Deregulating Cell Proliferation and Cell Cycle Control


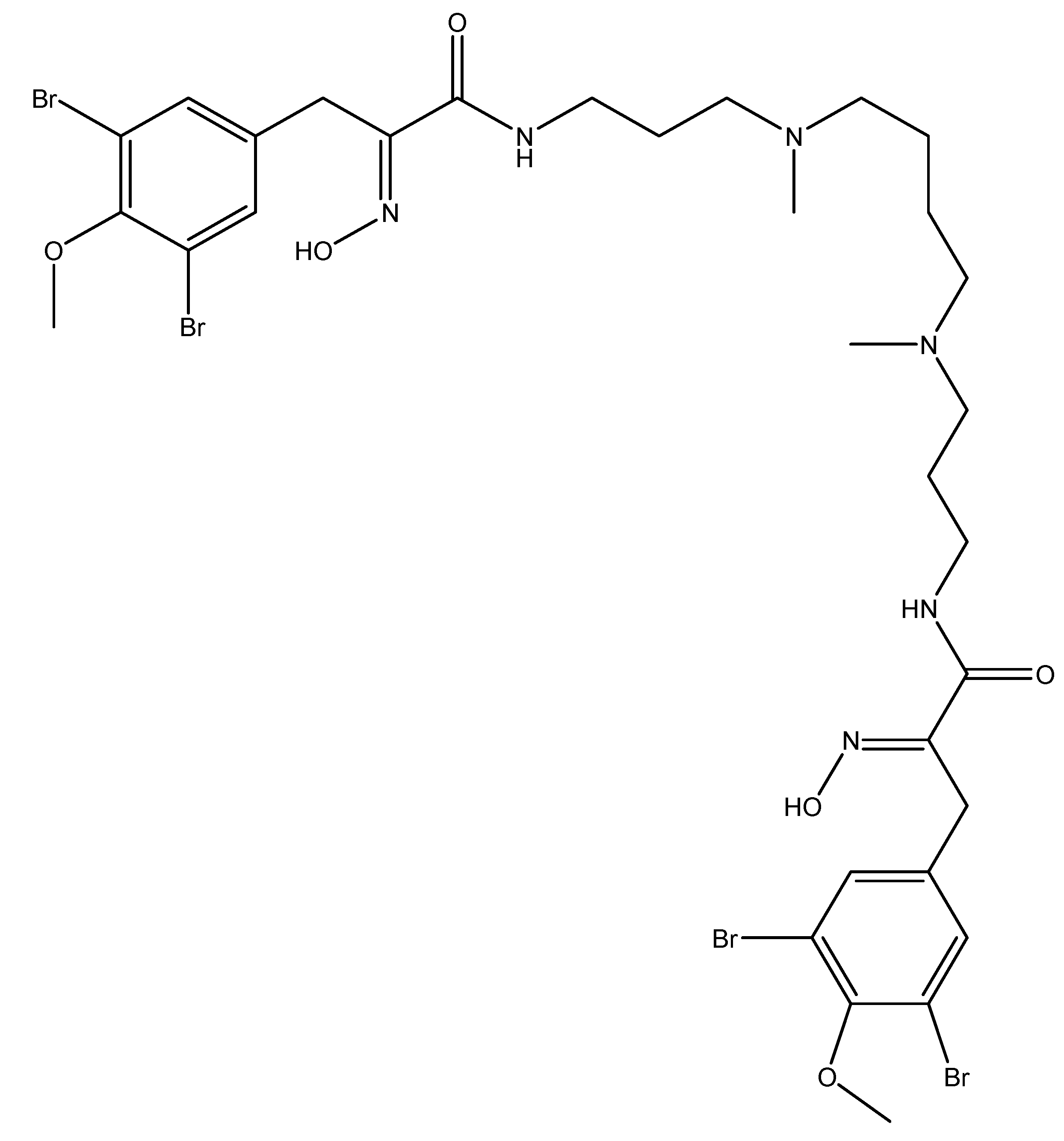
3.5. Targeting Angiogenesis


3.6. Targeting Isoprenylcysteine Carboxyl Methyltransferase

3.7. Cytotoxic Sponge-Derived Alkaloids with No Defined Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkaloid | Organism | Cell Lines | IC50 (μg/mL) | References |
|---|---|---|---|---|
| Amphimedine | Amphimedon sp. | U-87MG, U-373Mg, J82, HCT15, LoVo, A549 | 0.1–3.1 | [78] |
| Arenosclerin A–C | Arenosclera brasiliensis | HL60, B16, U138, L929 | <5 | [79] |
| Echinoclathrines A–C | Echinoclathria sp. | P388, A549m HT29 | 10 | [79] |
| Haliclonacyclamie E | Arenosclera brasiliensis | HL60, B16, U138, L929 | <5 | [79] |
| Halitulin | Haliclona tulearensis | P388, A549, MEL28 | 0.012–0.025 | [80] |
| Longamide | Agelas longissima, Homaxinella | P388 | Not determined | [81,82] |
| Ma’edamines A and B | Suberea sp. | L1210, KB | 3.9–5.2 | [83] |
| Matemone | Iotrochota purpurea | NSCLC-N6 L16, Mia PaCa-2, DU145 | 24–30 | [84] |
| N-Methyl-epi-manzamine D, epi-Manzamine D | PAL93055 | B16F10 | 0.1 | [85] |
| Nortopsentins A, B, and C | Spongsorites ruetzleri | P388 | 1.7–7.8 | [86] |
| Pyrinodemin A-D | Amphimedon sp. | L1210, KB | 0.06–0.08 | [87,88] |
| Topsentin B1 and B2 | Rhaphisia lacazei | NSCLC-N6 | 6.3–12 | [89] |
4. Tunicate-Derived Anti-Cancer Alkaloids: The Future Promise
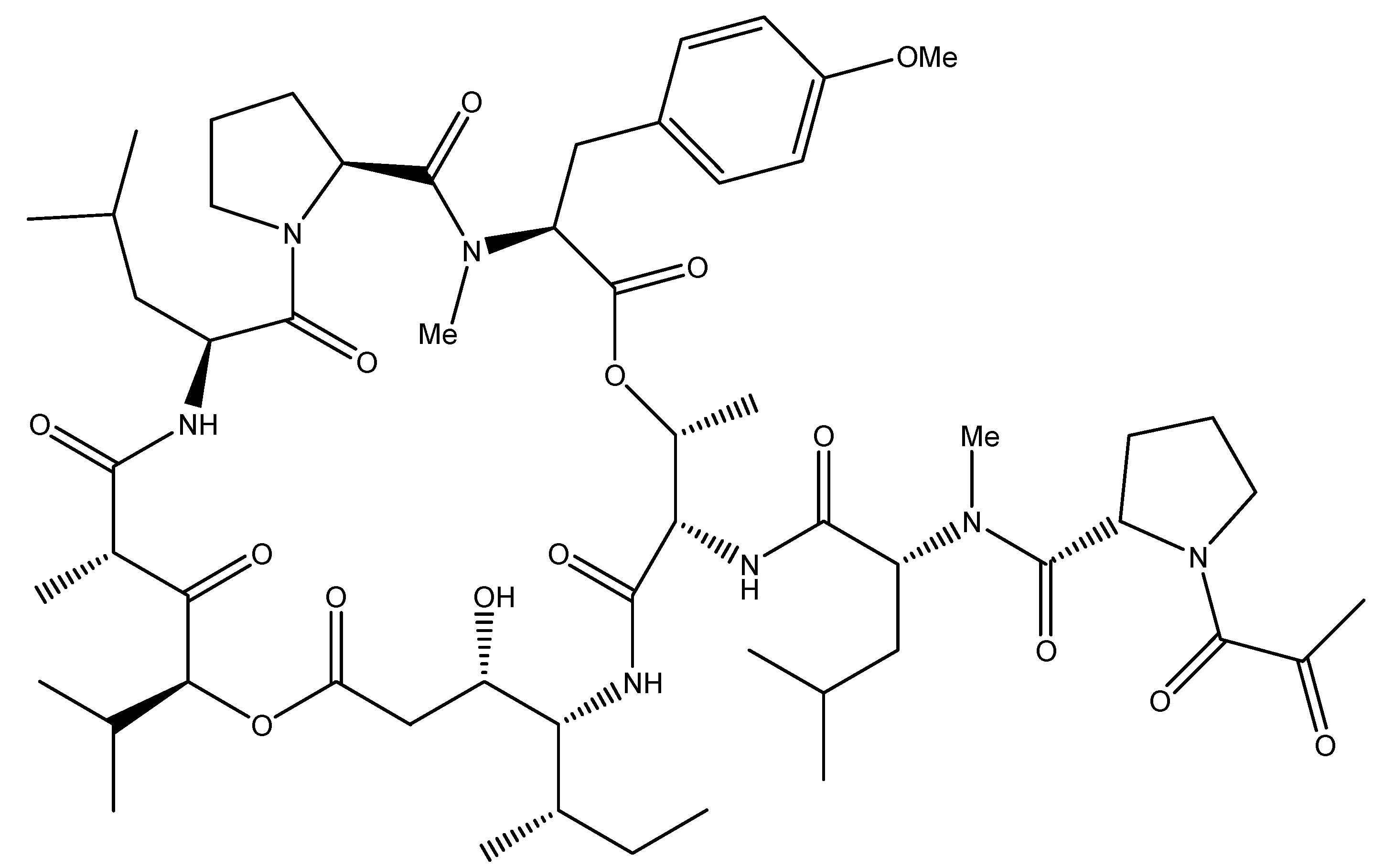
4.1. Aplidin: Oxidative-Stress Mediated Apoptosis

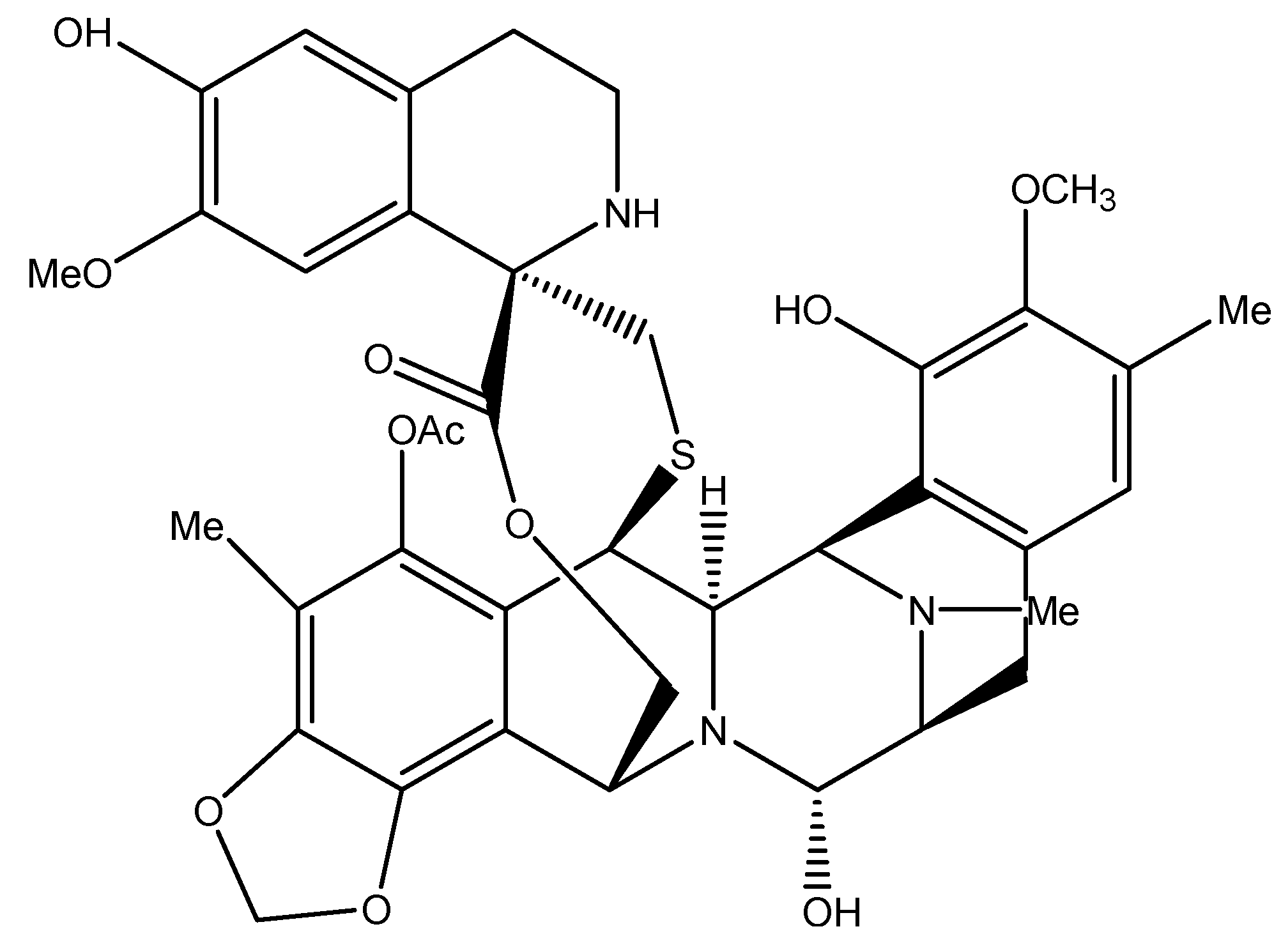
4.2. Trabectedin: A Unique Alkylating Agent

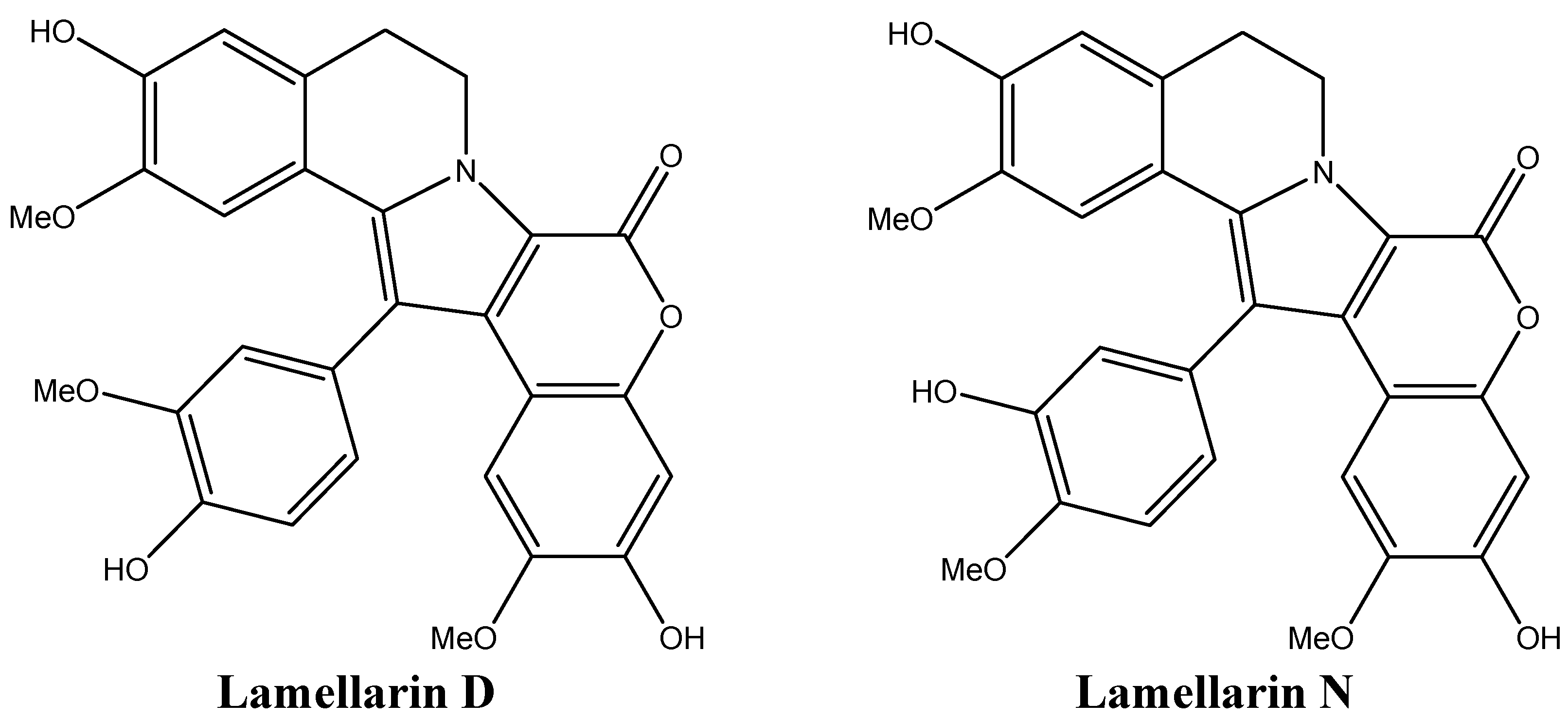
4.3. Lamellarin D: Overcoming Chemotherapy Resistance

4.4. Ascididemin: Topoisomerase II Inhibition

4.5. Lissoclinidine B: Stabilization of p53

4.6. Polycarpines: p53-Dependent Apoptosis

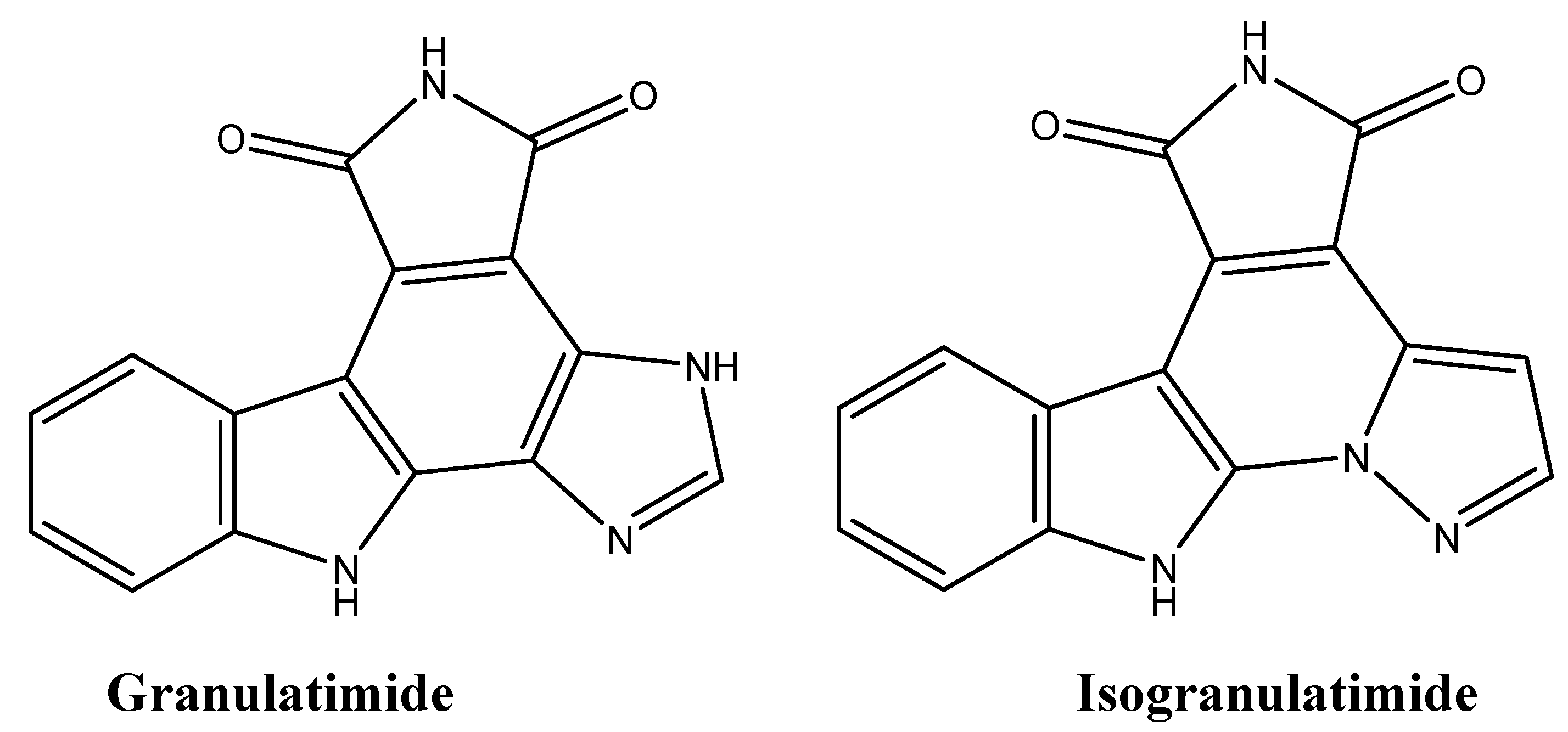
4.7. Granulatimide and Isogranulatimide: G2 Checkpoint Inhibition

4.8. Cytotoxic Tunicate-Derived Alkaloids with Undetermined Mechanisms of Action
| Alkaloid | Organism | Cell Lines | IC50(μg/mL) | References |
|---|---|---|---|---|
| Coproverdine | Tunicate | P388 | 0.95 | [127] |
| Eudistomins | Eudistoma gilboverde | LOX, OVCAR-3, COLO-205, MOLT-4 | <1.0 | [128] |
| Haouamine A | Aplidium haouarianum | HT-29 | 0.1 | [129] |
| Haterumaimide F | Lissoclinum voeltzkowi | P388 | 0.0055 | [130] |
| Kottamides A-D | Pycnoclavella kottae | P388 | >10 | [131] |
| Perophoramidine | Perophora namei | HCT116 | 60 | [132] |
| Pibocin B | Eudistoma sp. | Ehrlich carcinoma cells | Not determined | [133] |
| Sebastianines A and B | Cystodytes dellechiajei | HCT116 | <10 | [134] |
| Sulcatin | Microcosmus vulgaris | J774 | <10 | [135] |
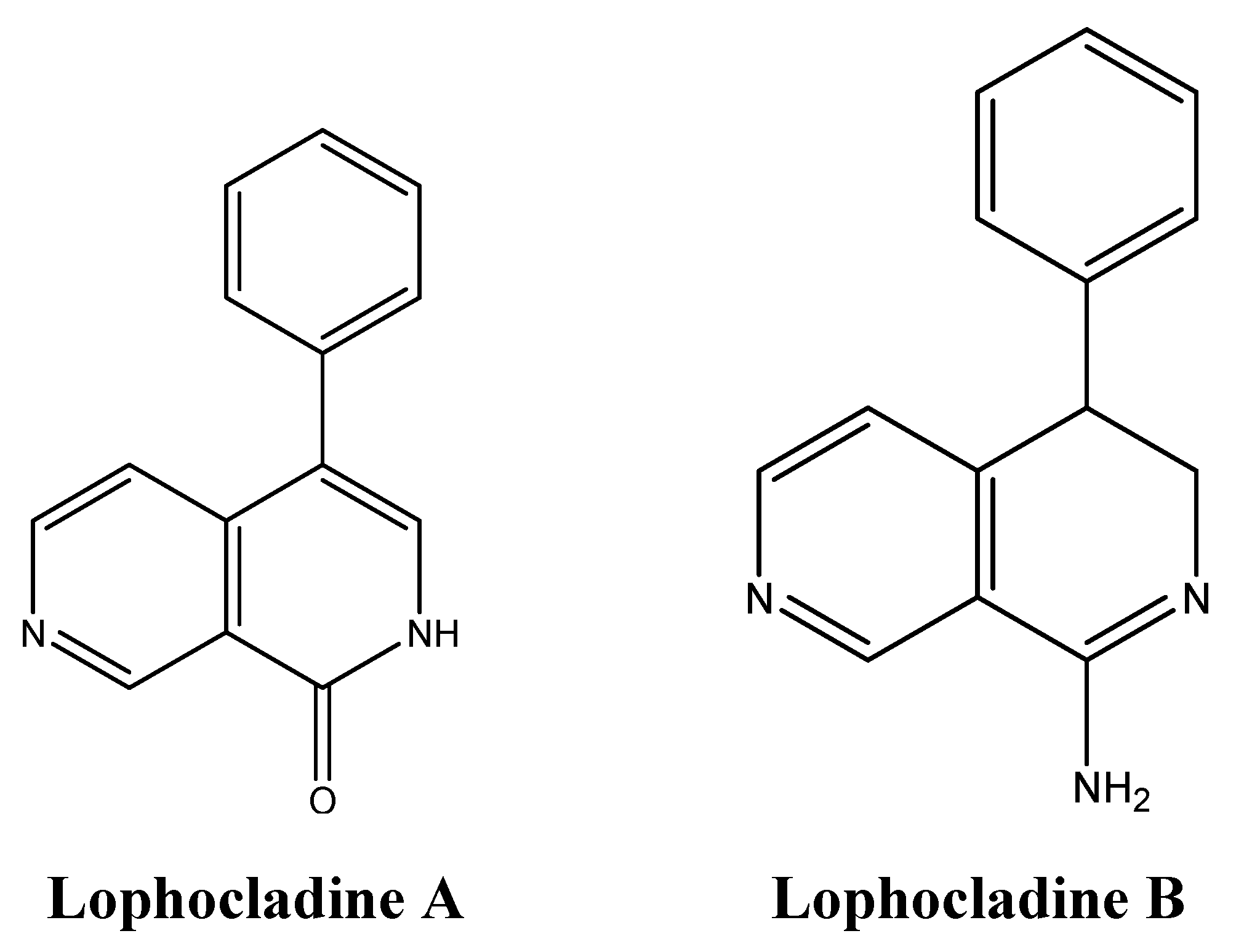
5. Anti-Cancer Alkaloids Derived from Algae: an Untapped Area of Study

6. Conclusions
| Alkaloid | Organism | Mechanisms of Action | References |
|---|---|---|---|
| Apratoxin | Cyanobacteria | G1 cell cycle arrest, inhibition of FGFR | [138] |
| Hectochlorin | Cyanobacteria | Hyperpolymerization of actin filaments | [10,11] |
| Largazole | Cyanobacteria | HDAC inhibitor | [20,21] |
| Lyngyabellin | Cyanobacteria | Hyperpolymerization of actin filaments | [14] |
| Shearinine E | Fungi | Inhibition of EGF | [25] |
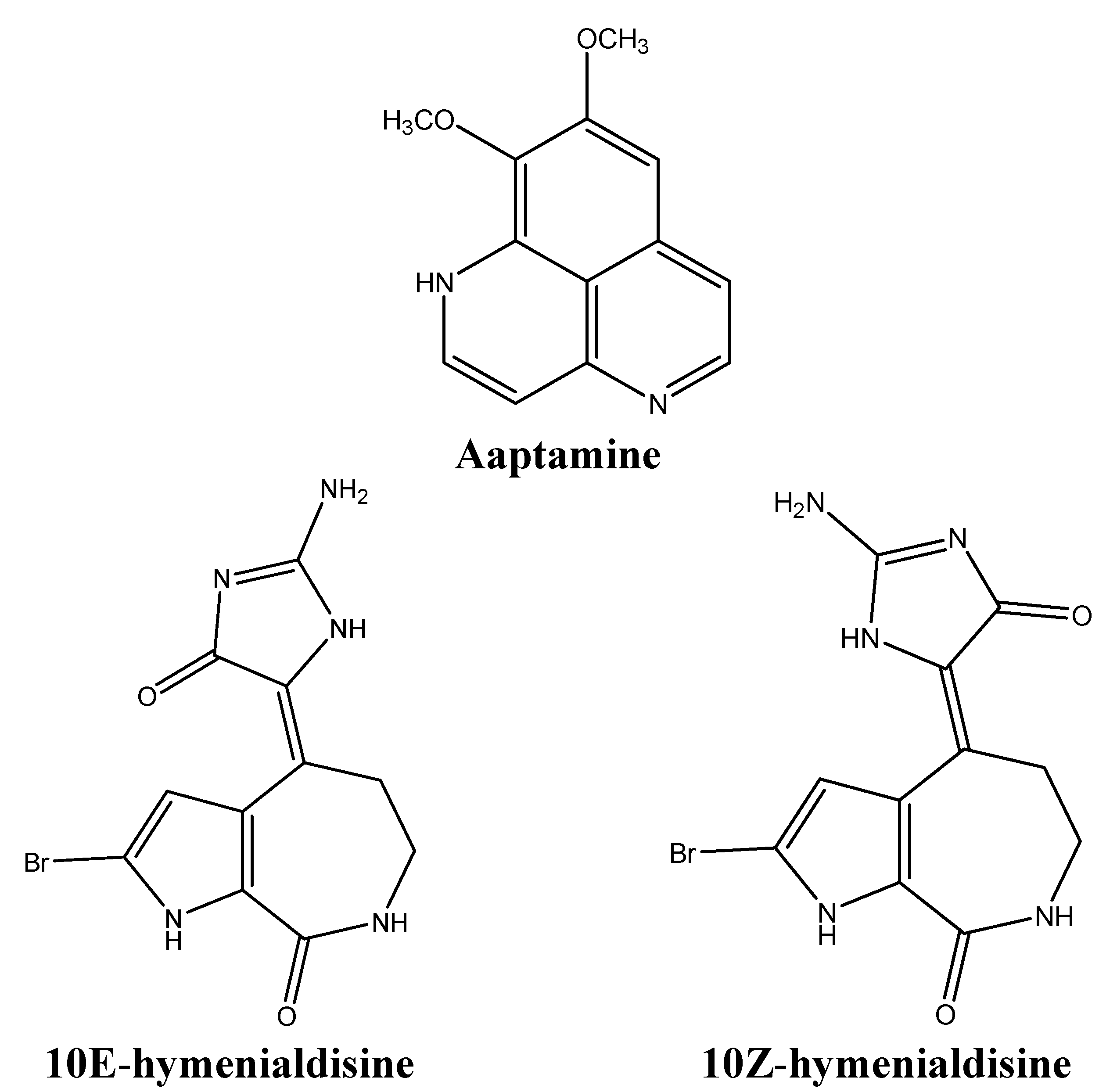
| Aaptamine | Sponges | ↑p27, G2/M cell cycle arrest | [66] |
| Aldisine alkaloids | Sponges | Inhibition of MEK-1, CDK1, Raf/MEK/MAPK | [67,68] |
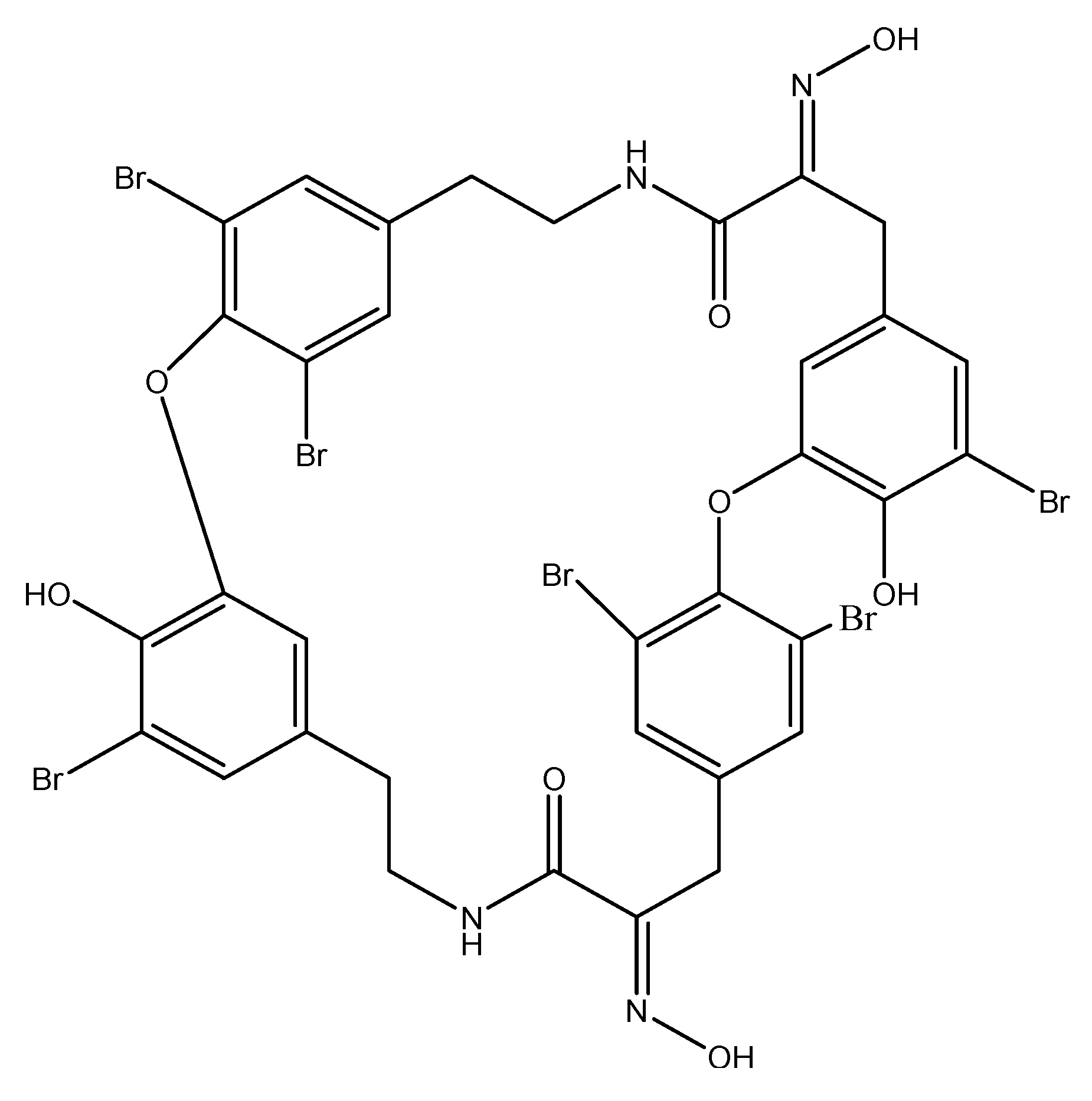
| Bastadin 6 | Sponges | Inhibition of VEGF and bFGF | [72] |
| E7974 | Sponges | G2/M cell cycle arrest, cleavage of caspase 3 and PARP, disruption of mitotic spindle formation | [43] |
| Hemiasterlin | Sponges | Tubulin depolymerization | [40] |
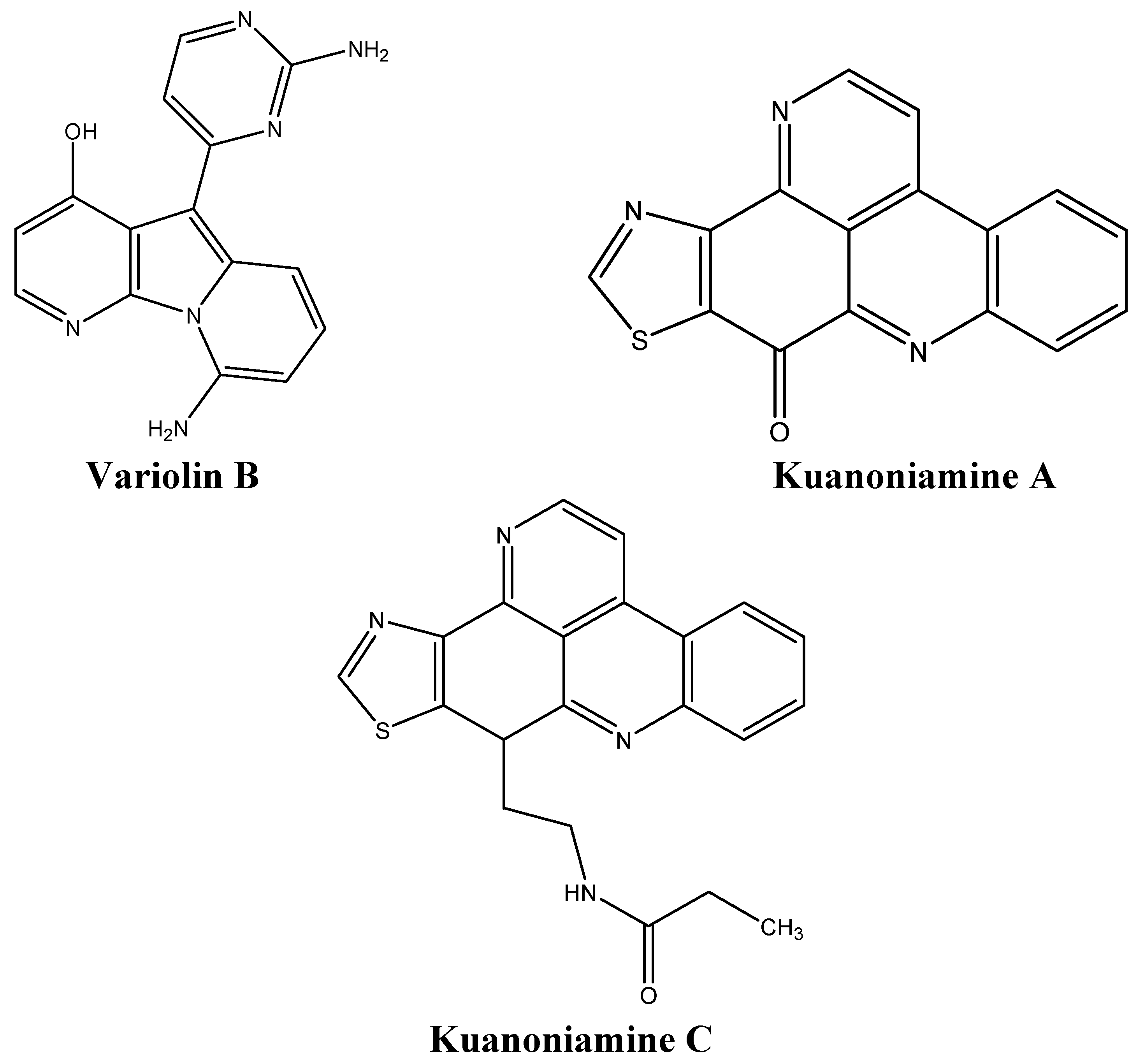
| Kuanoniamine A | Sponges | G1 cell cycle arrest | [70] |
| Makaluvamines | Sponges | Inhibition of TOP2 | [56] |
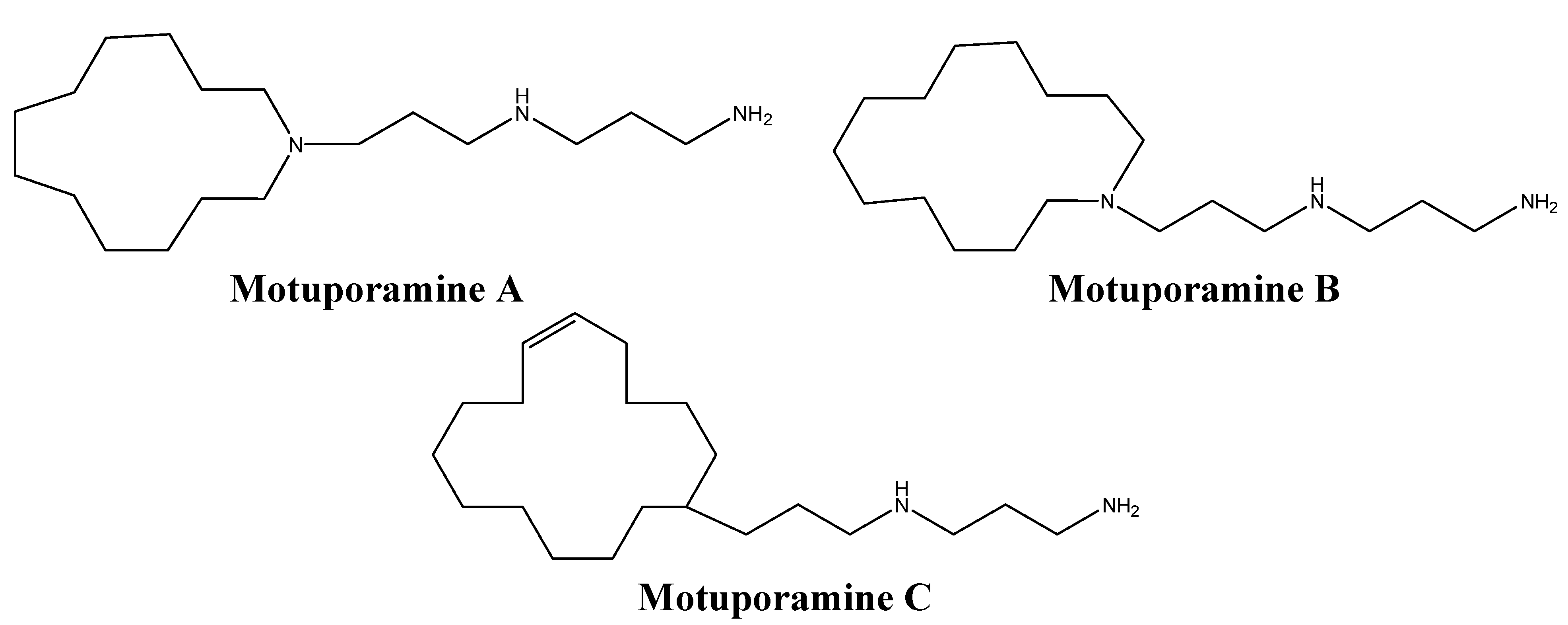
| Motuporamine C | Sponges | Inhibition of β1-integrin activation | [75] |
| Naamidine | Sponges | ↑p53, p21 Cdk↑, cleavage of capases 3, 8 and 9 and PARP | [61] |
| Neoamphimedine | Sponges | Inhibition of TOP2 | [49] |
| Renieramycin M | Sponges | ↑p53, Bcl-2↓, Mcl-1↓, Sensitization of cells to anoikis | [57] |
| Spermatinamine | Sponges | Inhibition of isoprenylcysteine carboxyl methyltransferase | [76] |
| Variolin B | Sponges | G1 and G2 cell cycle arrest | [139] |
| Aplidin | Tunicates | ↑p27, G1 cell cycle arrest, ↑ROS, ↑Src, ↑JNK, ↑p38MAPK, cytochrome c release, cleavage of caspases 3 and 9, PARP cleavage, ↓VEGF | [93,99,100] |
| Ascididemin | Tunicates | Inhibition of TOP2 | [122] |
| Granulatimide | Tunicates | Inhibition of G2 checkpoint | [126] |
| Lamellarin D | Tunicates | Inhibition of TOP1 and TOP2, cytochrome c release, cleavage of caspases 3 and 9, AIF translocation to the nucleus, ↑BAX | [116,120,121] |
| Lissoclinidine B | Tunicates | Inhibition of hdm2 | [123] |
| Polycarpines | Tunicates | ↑p53 | [125] |
| Trabectedin | Tunicates | DNA alkylation, S cell cycle arrest, RNA pol II breakdown, ↑CCL2, ↓VEGF, ↓IL-6 | [102] |
Acknowledgements
Potential Conflicts of Interest
References
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Morrison, W.B. Cancer chemotherapy: An annotated history. J. Vet. Intern. Med. 2010, 24, 1249–1262. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Munro, M.H.G.; Blunt, J.W.; Dumdei, E.J.; Hickford, S.J.; Lill, R.E.; Li, S.; Battershill, C.N.; Duckworth, A.R. The discovery and development of marine compounds with pharmaceutical potential. Prog. Ind. Microbiol. 1999, 35, 15–25. [Google Scholar] [CrossRef]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar]
- Weissman, K. Plumbing new depths in drug discovery. Chem. Biol. 2004, 11, 743–745. [Google Scholar] [CrossRef]
- Tan, L.T. Filamentous tropical marine cyanobacteria: A rich source of natural products for anticancer drugs. J. Appl. Phycol. 2010, 22, 659–676. [Google Scholar] [CrossRef]
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef]
- Ramaswamy, A.V.; Sorrels, C.M.; Gerwick, W.H. Cloning and biochemical characterization of the hectochlorin biosynthetic gene cluster from the marine cyanobacterium lyngbya majuscula. J. Nat. Prod. 2007, 70, 1977–1986. [Google Scholar] [CrossRef]
- Marquez, B.L.; Watts, K.S.; Yokochi, A.; Roberts, M.A.; Verdier-Pinard, P.; Jimenez, J.I.; Hamel, E.; Scheuer, P.J.; Gerwick, W.H. Structure and absolute stereochemistry of hectochlorin, a potent stimulator of actin assembly. J. Nat. Prod. 2002, 65, 866–871. [Google Scholar] [CrossRef]
- Han, B.; McPhail, K.L.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Isolation and structure of five lyngbyabellin derivatives from a papua new guinea collection of the marine cyanobacterium lyngbya majuscula. Tetrahedron 2005, 61, 11723–11729. [Google Scholar] [CrossRef]
- Yokokawa, F.; Sameshima, H.; Shioiri, T. Total synthesis of lyngbyabellin a, a potent cytotoxic metabolite from the marine cyanobacterium lyngbya majuscula. Tetrahedron Lett. 2001, 42, 4171–4174. [Google Scholar]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Mooberry, S.L. Isolation, structure determination, and biological activity of lyngbyabellin a from the marine cyanobacterium lyngbya majuscula. J. Nat. Prod. 2000, 63, 611–615. [Google Scholar]
- Milligan, K.E.; Marquez, B.L.; Williamson, R.T.; Gerwick, W.H. Lyngbyabellin b, a toxic and antifungal secondary metabolite from the marine cyanobacterium lyngbya majuscula. J. Nat. Prod. 2000, 63, 1440–1443. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Structurally diverse new alkaloids from palauan collections of the apratoxin-producing marine cyanobacterium lyngbya sp. Tetrahedron 2002, 58, 7959–7966. [Google Scholar] [CrossRef]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J.; Corbett, T.H. Total structure determination of apratoxin a, a potent novel cytotoxin from the marine cyanobacterium lyngbya majuscula. J. Am. Chem. Soc. 2001, 123, 5418–5423. [Google Scholar] [CrossRef]
- Chen, J.; Forsyth, C.J. Total synthesis of apratoxin a. J. Am. Chem. Soc. 2003, 125, 8734–8735. [Google Scholar] [CrossRef]
- Luesch, H.; Chanda, S.K.; Raya, R.M.; DeJesus, P.D.; Orth, A.P.; Walker, J.R.; Izpisúa Belmonte, J.C.; Schultz, P.G. A functional genomics approach to the mode of action of apratoxin A. Nat. Chem. Biol. 2006, 2, 158–167. [Google Scholar] [CrossRef]
- Cole, K.E.; Dowling, D.P.; Boone, M.A.; Phillips, A.J.; Christianson, D.W. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc. 2011, 133, 12474–12477. [Google Scholar]
- Wang, B.; Huang, P.H.; Chen, C.S.; Forsyth, C.J. Total syntheses of the histone deacetylase inhibitors largazole and 2-epi-largazole: Application of n-heterocyclic carbene mediated acylations in complex molecule synthesis. J. Org. Chem. 2011, 76, 1140–1150. [Google Scholar] [CrossRef]
- Pietra, F. Secondary metabolites from marine microorganisms: Bacteria, protozoa, algae and fungi. Achievements and prospects. Nat. Prod. Rep. 1997, 14, 453–464. [Google Scholar] [CrossRef]
- Liberra, K.; Lindequist, U. Marine fungi: A prolific resource of biologically active natural products? Pharmazie 1995, 50, 583–588. [Google Scholar]
- Liu, R.; Zhu, T.; Li, D.; Gu, J.; Xia, W.; Fang, Y.; Liu, Hongbing; Zhu, W.; Gu, Q. Two indolocarbazole alkaloids with apoptosis activity from a marine-derived actinomycete z(2)039-2. Arch. Pharm. Res. 2007, 30, 270–274. [Google Scholar] [CrossRef]
- Smetanina, O.F.; Kalinovsky, A.I.; Khudyakova, Y.V.; Pivkin, M.V.; Dmitrenok, P.S.; Fedorov, S.N.; Ji, H.; Kwak, J.Y.; Kuznetsova, T.A. Indole alkaloids produced by a marine fungus isolate of penicillium janthinellum biourge. J. Nat. Prod. 2007, 70, 906–909. [Google Scholar]
- Xin, Z.H.; Fang, Y.; Du, L.; Zhu, T.; Duan, L.; Chen, J.; Gu, Q.Q.; Zhu, W.M. Aurantiomides a-c, quinazoline alkaloids from the sponge-derived fungus penicillium aurantiogriseum sp0-19. J. Nat. Prod. 2007, 70, 853–855. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, W.L.; Fang, Y.C.; Zhu, T.J.; Gu, Q.Q.; Zhu, W.M. Cytotoxic alkaloids and antibiotic nordammarane triterpenoids from the marine-derived fungus aspergillus sydowi. J. Nat. Prod. 2008, 71, 985–989. [Google Scholar]
- Pawlik, J.R.; McFall, G.; Zea, S. Does the odor from sponges of the genus ircinia protect them from fish predators? J. Chem. Ecol. 2002, 28, 1103–1115. [Google Scholar] [CrossRef]
- Thomas, T.; Rusch, D.; DeMaere, M.Z.; Yung, P.Y.; Lewis, M.; Halpern, A.; Heidelberg, K.B.; Egan, S.; Steinberg, P.D.; Kjelleberg, S. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 2010, 4, 1557–1567. [Google Scholar] [CrossRef]
- Richelle-Maurer, E.; Gomez, R.; Braekman, J.C.; van de Vyver, G.; van Soest, R.W.; Devijver, C. Primary cultures from the marine sponge xestospongia muta (petrosiidae, haplosclerida). J. Biotechnol. 2003, 100, 169–176. [Google Scholar]
- Zhang, W.; Zhang, X.; Cao, X.; Xu, J.; Zhao, Q.; Yu, X.; Jin, M.; Deng, M. Optimizing the formation of in vitro sponge primmorphs from the chinese sponge stylotella agminata (ridley). J. Biotechnol. 2003, 100, 161–168. [Google Scholar]
- Belarbi, E.H.; Contreras Gómez, A.; Chisti, Y.; García Camacho, F.; Molina Grima, E. Producing drugs from marine sponges. Biotechnol. Adv. 2003, 21, 585–598. [Google Scholar] [CrossRef]
- Dembitsky, V.M.; Gloriozova, T.A.; Poroikov, V.V. Novel antitumor agents: Marine sponge alkaloids, their synthetic analogs and derivatives. Mini Rev. Med. Chem. 2005, 5, 319–336. [Google Scholar]
- Cigler, T.; Vahdat, L.T. Eribulin mesylate for the treatment of breast cancer. Expert Opin. Pharmacother. 2010, 11, 1587–1593. [Google Scholar] [CrossRef]
- Wade, R.H. On and around microtubules: An overview. Mol. Biotechnol. 2009, 43, 177–191. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Yamada, H.Y.; Rao, C.V. Genes that modulate the sensitivity for anti-microtubule drug-mediated chemotherapy. Curr. Cancer Drug Targets 2010, 10, 623–633. [Google Scholar] [CrossRef]
- Coleman, J.E.; Dilip de Silva, E.; Kong, F.; Andersen, R.J.; Allen, T.M. Cytotoxic peptides from the marine sponge cymbastela sp. Tetrahedron 1995, 51, 10653–10662. [Google Scholar]
- Talpir, R.; Benayahu, Y.; Kashman, Y.; Pannell, L.; Schleyer, M. Hemiasterlin and geodiamolide ta; two new cytotoxic peptides from the marine sponge hemiasterella minor (kirkpatrick). Tetrahedron Lett. 1994, 35, 4453–4456. [Google Scholar] [CrossRef]
- Anderson, H.J.; Coleman, J.E.; Andersen, R.J.; Roberge, M. Cytotoxic peptides hemiasterlin, hemiasterlin a and hemiasterlin b induce mitotic arrest and abnormal spindle formation. Cancer Chemother. Pharmacol. 1997, 39, 223–226. [Google Scholar]
- Jordan, M.A.; Thrower, D.; Wilson, L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 1992, 102, 401–416. [Google Scholar]
- Gamble, W.R.; Durso, N.A.; Fuller, R.W.; Westergaard, C.K.; Johnson, T.R.; Sackett, D.L.; Hamel, E.; Cardellina, J.H.; Boyd, M.R. Cytotoxic and tubulin-interactive hemiasterlins from auletta sp. and siphonochalina spp. Sponges. Bioorg. Med. Chem. 1999, 7, 1611–1615. [Google Scholar] [CrossRef]
- Kuznetsov, G.; TenDyke, K.; Towle, M.J.; Cheng, H.; Liu, J.; Marsh, J.P.; Schiller, S.E.; Spyvee, M.R.; Yang, H.; Seletsky, B.M.; et al. Tubulin-based antimitotic mechanism of e7974, a novel analogue of the marine sponge natural product hemiasterlin. Mol. Cancer Ther. 2009, 8, 2852–2860. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase i inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef]
- de Guzman, F.S.; Carte, B.; Troupe, N.; Faulkner, D.J.; Harper, M.K.; Concepcion, G.P.; Mangalindan, G.C.; Matsumoto, S.S.; Barrows, L.R.; Ireland, C.M. Cheminform abstract: Neoamphimedine: A new pyridoacridine topoisomerase ii inhibitor which catenates DNA. ChemInform 1999, 30. [Google Scholar] [CrossRef]
- Marshall, K.M.; Matsumoto, S.S.; Holden, J.A.; Concepción, G.P.; Tasdemir, D.; Ireland, C.M.; Barrows, L.R. The anti-neoplastic and novel topoisomerase ii-mediated cytotoxicity of neoamphimedine, a marine pyridoacridine. Biochem. Pharmacol. 2003, 66, 447–458. [Google Scholar]
- Low, R.L.; Kaguni, J.M.; Kornberg, A. Potent catenation of supercoiled and gapped DNA circles by topoisomerase i in the presence of a hydrophilic polymer. J. Biol. Chem. 1984, 259, 4576–4581. [Google Scholar]
- Holden, J.A.; Low, R.L. Characterization of a potent catenation activity of hela cell nuclei. J. Biol. Chem. 1985, 260, 14491–14497. [Google Scholar]
- Schmidt, E.W.; Harper, M.K.; Faulkner, D.J. Makaluvamines h-m and damirone c from the pohnpeian sponge zyzzya fuliginosa. J. Nat. Prod. 1995, 58, 1861–1867. [Google Scholar] [CrossRef]
- Hu, J.F.; Schetz, J.A.; Kelly, M.; Peng, J.N.; Ang, K.K.; Flotow, H.; Leong, C.Y.; Ng, S.B.; Buss, A.D.; Wilkins, S.P.; et al. New antiinfective and human 5-ht2 receptor binding natural and semisynthetic compounds from the jamaican sponge smenospongia aurea. J. Nat. Prod. 2002, 65, 476–480. [Google Scholar] [CrossRef]
- Carney, J.R.; Scheuer, P.J.; Kelly-Borges, M. A new bastadin from the sponge psammaplysilla purpurea. J. Nat. Prod. 1993, 56, 153–157. [Google Scholar] [CrossRef]
- Barrows, L.R.; Radisky, D.C.; Copp, B.R.; Swaffar, D.S.; Kramer, R.A.; Warters, R.L.; Ireland, C.M. Makaluvamines, marine natural products, are active anti-cancer agents and DNA topo ii inhibitors. Anticancer Drug Des. 1993, 8, 333–347. [Google Scholar]
- Shinkre, B.A.; Raisch, K.P.; Fan, L.; Velu, S.E. Analogs of the marine alkaloid makaluvamines: Synthesis, topoisomerase ii inhibition, and anticancer activity. Bioorg. Med. Chem. Lett. 2007, 17, 2890–2893. [Google Scholar] [CrossRef]
- Halim, H.; Chunhacha, P.; Suwanborirux, K.; Chanvorachote, P. Anticancer and antimetastatic activities of renieramycin m, a marine tetrahydroisoquinoline alkaloid, in human non-small cell lung cancer cells. Anticancer Res. 2011, 31, 193–201. [Google Scholar]
- Saito, N.; Tanaka, C.; Koizumi, Y.-i.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of renieramycin m into jorumycin and renieramycin j including oxidative degradation products, mimosamycin, renierone, and renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Nimmannit, U.; Stehlik, C.; Wang, L.; Jiang, B.H.; Ongpipatanakul, B.; Rojanasakul, Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through s-nitrosylation and inhibition of bcl-2 ubiquitination. Cancer Res. 2006, 66, 6353–6360. [Google Scholar] [CrossRef]
- Carmelya, S.; Ilanb, M.; Kashmana, Y. 2-amino imidazole alkaloids from the marine sponge leucetta chagosensis. Tetrahedron 1989, 45, 2193–2200. [Google Scholar] [CrossRef]
- LaBarbera, D.V.; Modzelewska, K.; Glazar, A.I.; Gray, P.D.; Kaur, M.; Liu, T.; Grossman, D.; Harper, M.K.; Kuwada, S.K.; Moghal, N.; et al. The marine alkaloid naamidine a promotes caspase-dependent apoptosis in tumor cells. Anticancer Drugs 2009, 20, 425–436. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. Waf1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 cdk-interacting protein cip1 is a potent inhibitor of g1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Aoki, S.; Kong, D.; Suna, H.; Sowa, Y.; Sakai, T.; Setiawan, A.; Kobayashi, M. Aaptamine, a spongean alkaloid, activates p21 promoter in a p53-independent manner. Biochem. Biophys. Res. Commun. 2006, 342, 101–106. [Google Scholar] [CrossRef]
- Tasdemir, D.; Mallon, R.; Greenstein, M.; Feldberg, L.R.; Kim, S.C.; Collins, K.; Wojciechowicz, D.; Mangalindan, G.C.; Concepción, G.P.; Harper, M.K.; et al. Aldisine alkaloids from the philippine sponge stylissa massa are potent inhibitors of mitogen-activated protein kinase kinase-1 (mek-1). J. Med. Chem. 2002, 45, 529–532. [Google Scholar] [CrossRef]
- Meijer, L.; Thunnissen, A.M.; White, A.W.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, gsk-3beta and ck1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Erba, E.; Serafini, M.; Gaipa, G.; Tognon, G.; Marchini, S.; Celli, N.; Rotilio, D.; Broggini, M.; Jimeno, J.; Faircloth, G.T.; et al. Effect of aplidin in acute lymphoblastic leukaemia cells. Br. J. Cancer 2003, 89, 763–773. [Google Scholar] [CrossRef]
- Kijjoa, A.; Wattanadilok, R.; Campos, N.; Nascimento, M.S.; Pinto, M.; Herz, W. Anticancer activity evaluation of kuanoniamines a and c isolated from the marine sponge oceanapia sagittaria, collected from the gulf of thailand. Mar. Drugs 2007, 5, 6–22. [Google Scholar] [CrossRef]
- Prager, G.W.; Poettler, M. Angiogenesis in cancer. Basic mechanisms and therapeutic advances. Hamostaseologie 2012, 32. [Google Scholar] [CrossRef]
- Aoki, S.; Cho, S.H.; Ono, M.; Kuwano, T.; Nakao, S.; Kuwano, M.; Nakagawa, S.; Gao, J.Q.; Mayumi, T.; Shibuya, M.; et al. Bastadin 6, a spongean brominated tyrosine derivative, inhibits tumor angiogenesis by inducing selective apoptosis to endothelial cells. Anticancer Drugs 2006, 17, 269–278. [Google Scholar] [CrossRef]
- Goldring, W.P.; Weiler, L. Cytotoxic alkaloids motuporamines a-c: Synthesis and structural verification. Org. Lett. 1999, 1, 1471–1473. [Google Scholar] [CrossRef]
- Roskelley, C.D.; Williams, D.E.; McHardy, L.M.; Leong, K.G.; Troussard, A.; Karsan, A.; Andersen, R.J.; Dedhar, S.; Roberge, M. Inhibition of tumor cell invasion and angiogenesis by motuporamines. Cancer Res. 2001, 61, 6788–6794. [Google Scholar]
- Williams, D.E.; Craig, K.S.; Patrick, B.; McHardy, L.M.; van Soest, R.; Roberge, M.; Andersen, R.J. Motuporamines, anti-invasion and anti-angiogenic alkaloids from the marine sponge xestospongia exigua (kirkpatrick): Isolation, structure elucidation, analogue synthesis, and conformational analysis. J. Org. Chem. 2002, 67, 245–258. [Google Scholar] [CrossRef]
- Buchanan, M.S.; Carroll, A.R.; Fechner, G.A.; Boyle, A.; Simpson, M.M.; Addepalli, R.; Avery, V.M.; Hooper, J.N.; Su, N.; Chen, H.; et al. Spermatinamine, the first natural product inhibitor of isoprenylcysteine carboxyl methyltransferase, a new cancer target. Bioorg. Med. Chem. Lett. 2007, 17, 6860–6863. [Google Scholar]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef]
- Brahic, C.; Darro, F.; Belloir, M.; Bastide, J.; Kiss, R.; Delfourne, E. Synthesis and cytotoxic evaluation of analogues of the marine pyridoacridine amphimedine. Bioorg. Med. Chem. 2002, 10, 2845–2853. [Google Scholar] [CrossRef]
- Torres, Y.R.; Berlinck, R.G.; Magalhães, A.; Schefer, A.B.; Ferreira, A.G.; Hajdu, E.; Muricy, G. Arenosclerins a-c and haliclonacyclamine e, new tetracyclic alkaloids from a brazilian endemic haplosclerid sponge arenosclera brasiliensis. J. Nat. Prod. 2000, 63, 1098–1105. [Google Scholar] [CrossRef]
- Kashman, Y.; Koren-Goldshlager, G.; Gravalos, M.D.G.; Schleyer, M. Halitulin, a new cytotoxic alkaloid from the marine sponge haliclona tulearensis. Tetrahedron Lett. 1999, 40, 997–1000. [Google Scholar] [CrossRef]
- Cafieri, F.; Fattorusso, E.; Mangoni, A.; Taglialatela-Scafati, O. Longamide and 3,7-dimethylisoguanine, two novel alkaloids from the marine sponge agelas longissima. Tetrahedron Lett. 1995, 36, 7893–7896. [Google Scholar] [CrossRef]
- Umeyama, A.; Ito, S.; Yuasa, E.; Arihara, S.; Yamada, T. A new bromopyrrole alkaloid and the optical resolution of the racemate from the marine sponge homaxinella sp. J. Nat. Prod. 1998, 61, 1433–1434. [Google Scholar] [CrossRef]
- Hirano, K.; Kubota, T.; Tsuda, M.; Watanabe, K.; Fromont, J.; Kobayashi, J.I. Ma'edamines a and b, cytotoxic bromotyrosine alkaloids with a unique 2(1h)pyrazinone ring from sponge suberea sp. Tetrahedron 2000, 56, 8107–8110. [Google Scholar] [CrossRef]
- Carletti, I.; Banaigs, B.; Amade, P. Matemone, a new bioactive bromine-containing oxindole alkaloid from the indian ocean sponge iotrochota purpurea. J. Nat. Prod. 2000, 63, 981–983. [Google Scholar] [CrossRef]
- Zhou, B.-N.; Slebodnick, C.; Johnson, R.K.; Mattern, M.R.; Kingston, D.G.I. New cytotoxic manzamine alkaloids from a palaun sponge. Tetrahedron 2000, 56, 5781–5784. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Romeril, S.P.; Lee, V.; Claridge, T.D. Studies toward the total synthesis of the cytotoxic sponge alkaloid pyrinodemin a. Org. Lett. 2001, 3, 1145–1148. [Google Scholar] [CrossRef]
- Hirano, K.; Kubota, T.; Tsuda, M.; Mikami, Y.; Kobayashi, J. Pyrinodemins b-d, potent cytotoxicbis-pyridine alkaloids from marine sponge amphimedon sp. Chem. Pharm. Bull. 2000, 48, 974–977. [Google Scholar] [CrossRef]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New bisindole alkaloids of the topsentin and hamacanthin classes from the mediterranean marine sponge rhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef]
- Zubia, E.; Ortega, M.J.; Salva, J. Natural products chemistry in marine ascidians of the genus aplidium. MiniRev. Org. Chem. 2005, 2, 389–399. [Google Scholar] [CrossRef]
- Rinehart, K.L. Antitumor compounds from tunicates. Med. Res. Rev. 2000, 20, 1–27. [Google Scholar] [CrossRef]
- Rockwell, S.; Liu, Y. Aplidin as a potential adjunct to radiation therapy: In vitro studies. Int. J. Radiat. Biol. 2010, 86, 63–70. [Google Scholar] [CrossRef]
- Cuadrado, A.; Garcia-Fernandez, L.F.; Gonzalez, L.; Suarez, Y.; Losada, A.; Alcaide, V.; Martinez, T.; Fernandez-Sousa, J.M.; Sanchez-Puelles, J.M.; Munoz, A. Aplidin induces apoptosis in human cancer cells via glutathione depletion and sustained activation of the epidermal growth factor receptor, src, jnk, and p38 mapk. J. Biol. Chem. 2003, 278, 241–250. [Google Scholar]
- González-Santiago, L.; Suárez, Y.; Zarich, N.; Muñoz-Alonso, M.J.; Cuadrado, A.; Martínez, T.; Goya, L.; Iradi, A.; Sáez-Tormo, G.; Maier, J.V.; et al. Aplidin induces jnk-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, rac1 gtpase activation, and mkp-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar]
- García-Fernández, L.F.; Losada, A.; Alcaide, V.; Alvarez, A.M.; Cuadrado, A.; González, L.; Nakayama, K.; Nakayama, K.I.; Fernández-Sousa, J.M.; Muñoz, A.; et al. Aplidin induces the mitochondrial apoptotic pathway via oxidative stress-mediated jnk and p38 activation and protein kinase c delta. Oncogene 2002, 21, 7533–7544. [Google Scholar] [CrossRef]
- Gajate, C.; An, F.; Mollinedo, F. Rapid and selective apoptosis in human leukemic cells induced by aplidine through a fas/cd95- and mitochondrial-mediated mechanism. Clin. Cancer Res. 2003, 9, 1535–1545. [Google Scholar]
- Taraboletti, G.; Poli, M.; Dossi, R.; Manenti, L.; Borsotti, P.; Faircloth, G.T.; Broggini, M.; D'Incalci, M.; Ribatti, D.; Giavazzi, R. Antiangiogenic activity of aplidine, a new agent of marine origin. Br. J. Cancer 2004, 90, 2418–2424. [Google Scholar]
- Biscardi, M.; Caporale, R.; Balestri, F.; Gavazzi, S.; Jimeno, J.; Grossi, A. Vegf inhibition and cytotoxic effect of aplidin in leukemia cell lines and cells from acute myeloid leukemia. Ann. Oncol. 2005, 16, 1667–1674. [Google Scholar] [CrossRef]
- Broggini, M.; Marchini, S.V.; Galliera, E.; Borsotti, P.; Taraboletti, G.; Erba, E.; Sironi, M.; Jimeno, J.; Faircloth, G.T.; Giavazzi, R.; et al. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (vegf) secretion and blocks vegf-vegfr-1 (flt-1) autocrine loop in human leukemia cells molt-4. Leukemia 2003, 17, 52–59. [Google Scholar] [CrossRef]
- Moneo, V.; Serelde, B.G.; Leal, J.F.; Blanco-Aparicio, C.; Diaz-Uriarte, R.; Aracil, M.; Tercero, J.C.; Jimeno, J.; Carnero, A. Levels of p27(kip1) determine aplidin sensitivity. Mol. Cancer Ther. 2007, 6, 1310–1316. [Google Scholar] [CrossRef]
- Trabectedin: Ecteinascidin 743, ecteinascidin-743, et 743, et-743, nsc 684766. Drugs R D 2006, 7, 317–328. [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (et-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef]
- Hurley, L.H.; Zewail-Foote, M. The antitumor agent ecteinascidin 743: Characterization of its covalent DNA adducts and chemical stability. Adv. Exp. Med. Biol. 2001, 500, 289–299. [Google Scholar]
- Herrero, A.B.; Martín-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-talk between nucleotide excision and homologous recombination DNA repair pathways in the mechanism of action of antitumor trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef]
- Soares, D.G.; Escargueil, A.E.; Poindessous, V.; Sarasin, A.; de Gramont, A.; Bonatto, D.; Henriques, J.A.; Larsen, A.K. Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc. Natl. Acad. Sci. USA 2007, 104, 13062–13067. [Google Scholar]
- Tavecchio, M.; Simone, M.; Erba, E.; Chiolo, I.; Liberi, G.; Foiani, M.; D’Incalci, M.; Damia, G. Role of homologous recombination in trabectedin-induced DNA damage. Eur. J. Cancer 2008, 44, 609–618. [Google Scholar] [CrossRef]
- Scotto, K.W. Et-743: More than an innovative mechanism of action. Anticancer Drugs 2002, 13 (Suppl. 1), S3–S6.
- Minuzzo, M.; Marchini, S.; Broggini, M.; Faircloth, G.; D’Incalci, M.; Mantovani, R. Interference of transcriptional activation by the antineoplastic drug ecteinascidin-743. Proc. Natl. Acad. Sci. USA 2000, 97, 6780–6784. [Google Scholar]
- Minuzzo, M.; Ceribelli, M.; Pitarque-Martì, M.; Borrelli, S.; Erba, E.; DiSilvio, A.; D’Incalci, M.; Mantovani, R. Selective effects of the anticancer drug yondelis (et-743) on cell-cycle promoters. Mol. Pharmacol. 2005, 68, 1496–1503. [Google Scholar] [CrossRef]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits mdr1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef]
- Friedman, D.; Hu, Z.; Kolb, E.A.; Gorfajn, B.; Scotto, K.W. Ecteinascidin-743 inhibits activated but not constitutive transcription. Cancer Res. 2002, 62, 3377–3381. [Google Scholar]
- Aune, G.J.; Takagi, K.; Sordet, O.; Guirouilh-Barbat, J.; Antony, S.; Bohr, V.A.; Pommier, Y. Von hippel-lindau-coupled and transcription-coupled nucleotide excision repair-dependent degradation of rna polymerase ii in response to trabectedin. Clin. Cancer Res. 2008, 14, 6449–6455. [Google Scholar] [CrossRef]
- Allavena, P.; Signorelli, M.; Chieppa, M.; Erba, E.; Bianchi, G.; Marchesi, F.; Olimpio, C.O.; Bonardi, C.; Garbi, A.; Lissoni, A.; et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): Inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005, 65, 2964–2971. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar]
- Sessa, C.; D’Incalci, M. Trabectedin in ovarian cancer: Could we expect more? Ann. Oncol. 2011, 22, 7–8. [Google Scholar] [CrossRef]
- Pla, D.; Albericio, F.; Alvarez, M. Recent advances in lamellarin alkaloids: Isolation, synthesis and activity. Anticancer Agents Med. Chem. 2008, 8, 746–760. [Google Scholar]
- Reddy, A.V.; Ravinder, K.; Narasimhulu, M.; Sridevi, A.; Satyanarayana, N.; Kondapi, A.K.; Venkateswarlu, Y. New anticancer bastadin alkaloids from the sponge dendrilla cactos. Bioorg. Med. Chem. 2006, 14, 4452–4457. [Google Scholar] [CrossRef]
- Marco, E.; Laine, W.; Tardy, C.; Lansiaux, A.; Iwao, M.; Ishibashi, F.; Bailly, C.; Gago, F. Molecular determinants of topoisomerase i poisoning by lamellarins: Comparison with camptothecin and structure-activity relationships. J. Med. Chem. 2005, 48, 3796–3807. [Google Scholar] [CrossRef]
- Kluza, J.; Gallego, M.A.; Loyens, A.; Beauvillain, J.C.; Sousa-Faro, J.M.; Cuevas, C.; Marchetti, P.; Bailly, C. Cancer cell mitochondria are direct proapoptotic targets for the marine antitumor drug lamellarin d. Cancer Res. 2006, 66, 3177–3187. [Google Scholar]
- Gallego, M.A.; Ballot, C.; Kluza, J.; Hajji, N.; Martoriati, A.; Castéra, L.; Cuevas, C.; Formstecher, P.; Joseph, B.; Kroemer, G.; et al. Overcoming chemoresistance of non-small cell lung carcinoma through restoration of an aif-dependent apoptotic pathway. Oncogene 2008, 27, 1981–1992. [Google Scholar] [CrossRef]
- Baunbaek, D.; Trinkler, N.; Ferandin, Y.; Lozach, O.; Ploypradith, P.; Rucirawat, S.; Ishibashi, F.; Iwao, M.; Meijer, L. Anticancer alkaloid lamellarins inhibit protein kinases. Mar. Drugs 2008, 6, 514–527. [Google Scholar] [CrossRef]
- Dassonneville, L.; Wattez, N.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Banaigs, B.; Bonnard, I.; Bailly, C. Inhibition of topoisomerase ii by the marine alkaloid ascididemin and induction of apoptosis in leukemia cells. Biochem. Pharmacol. 2000, 60, 527–537. [Google Scholar]
- Martinez, J.D. Restoring p53 tumor suppressor activity as an anticancer therapeutic strategy. Future Oncol. 2010, 6, 1857–1862. [Google Scholar] [CrossRef]
- Clement, J.A.; Kitagaki, J.; Yang, Y.; Saucedo, C.J.; O'Keefe, B.R.; Weissman, A.M.; McKee, T.C.; McMahon, J.B. Discovery of new pyridoacridine alkaloids from lissoclinum cf. Badium that inhibit the ubiquitin ligase activity of hdm2 and stabilize p53. Bioorg. Med. Chem. 2008, 16, 10022–10028. [Google Scholar]
- Fedorov, S.N.; Bode, A.M.; Stonik, V.A.; Gorshkova, I.A.; Schmid, P.C.; Radchenko, O.S.; Berdyshev, E.V.; Dong, Z. Marine alkaloid polycarpine and its synthetic derivative dimethylpolycarpine induce apoptosis in jb6 cells through p53- and caspase 3-dependent pathways. Pharm. Res. 2004, 21, 2307–2319. [Google Scholar] [CrossRef]
- Berlinck, R.G.S.; Britton, R.; Piers, E.; Lim, L.; Roberge, M.; Moreira da Rocha, R.; Andersen, R.J. Granulatimide and isogranulatimide, aromatic alkaloids with g2 checkpoint inhibition activity isolated from the brazilian ascidian didemnum granulatum: Structure elucidation and synthesis. J. Org. Chem. 1998, 63, 9850–9856. [Google Scholar]
- Urban, S.; Blunt, J.W.; Munro, M.H. Coproverdine, a novel, cytotoxic marine alkaloid from a new zealand ascidian. J. Nat. Prod. 2002, 65, 1371–1373. [Google Scholar] [CrossRef]
- Rashid, M.A.; Gustafson, K.R.; Boyd, M.R. New cytotoxic n-methylated beta-carboline alkaloids from the marine ascidian eudistoma gilboverde. J. Nat. Prod. 2001, 64, 1454–1456. [Google Scholar] [CrossRef]
- Garrido, L.; Zubía, E.; Ortega, M.J.; Salvá, J. Haouamines a and b: A new class of alkaloids from the ascidian aplidium haouarianum. J. Org. Chem. 2003, 68, 293–299. [Google Scholar] [CrossRef]
- Uddin, M.J.; Kokubo, S.; Ueda, K.; Suenaga, K.; Uemura, D. Haterumaimides f-i, four new cytotoxic diterpene alkaloids from an ascidian lissoclinum species. J. Nat. Prod. 2001, 64, 1169–1173. [Google Scholar] [CrossRef]
- Appleton, D.R.; Page, M.J.; Lambert, G.; Berridge, M.V.; Copp, B.R. Kottamides a-d: Novel bioactive imidazolone-containing alkaloids from the new zealand ascidian pycnoclavella kottae. J. Org. Chem. 2002, 67, 5402–5404. [Google Scholar]
- Verbitski, S.M.; Mayne, C.L.; Davis, R.A.; Concepcion, G.P.; Ireland, C.M. Isolation, structure determination, and biological activity of a novel alkaloid, perophoramidine, from the philippine ascidian perophora namei. J. Org. Chem. 2002, 67, 7124–7126. [Google Scholar] [CrossRef]
- Makarieva, T.N.; Dmitrenok, A.S.; Dmitrenok, P.S.; Grebnev, B.B.; Stonik, V.A. Pibocin b, the first n-o-methylindole marine alkaloid, a metabolite from the far-eastern ascidian eudistoma species. J. Nat. Prod. 2001, 64, 1559–1561. [Google Scholar] [CrossRef]
- Torres, Y.R.; Bugni, T.S.; Berlinck, R.G.; Ireland, C.M.; Magalhães, A.; Ferreira, A.G.; Moreira Da Rocha, R. Sebastianines a and b, novel biologically active pyridoacridine alkaloids from the brazilian ascidian cystodytes dellechiajei. J. Org. Chem. 2002, 67, 5429–5432. [Google Scholar]
- Aiello, A.; Fattorusso, E.; Menna, M.; Iuvone, T. Sulcatin, a novel antiproliferative n-methylpyridinium alkaloid from the ascidian microcosmus vulgaris. J. Nat. Prod. 2000, 63, 517–519. [Google Scholar] [CrossRef]
- Güven, K.C.; Percot, A.; Sezik, E. Alkaloids in marine algae. Mar. Drugs 2010, 8, 269–284. [Google Scholar] [CrossRef]
- Gross, H.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Ballantine, D.L.; Murray, T.F.; Valeriote, F.A.; Gerwick, W.H. Lophocladines, bioactive alkaloids from the red alga lophocladia sp. J. Nat. Prod. 2006, 69, 640–644. [Google Scholar] [CrossRef]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar] [CrossRef]
- Bergamaschi, D.; Ronzoni, S.; Taverna, S.; Faretta, M.; De Feudis, P.; Faircloth, G.; Jimeno, J.; Erba, E.; D'Incalci, M. Cell cycle perturbations and apoptosis induced by isohomohalichondrin b (ihb), a natural marine compound. Br. J. Cancer 1999, 79, 267–277. [Google Scholar] [CrossRef]
- Samples Availability: Not available.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tohme, R.; Darwiche, N.; Gali-Muhtasib, H. A Journey Under the Sea: The Quest for Marine Anti-Cancer Alkaloids. Molecules 2011, 16, 9665-9696. https://doi.org/10.3390/molecules16119665
Tohme R, Darwiche N, Gali-Muhtasib H. A Journey Under the Sea: The Quest for Marine Anti-Cancer Alkaloids. Molecules. 2011; 16(11):9665-9696. https://doi.org/10.3390/molecules16119665
Chicago/Turabian StyleTohme, Rita, Nadine Darwiche, and Hala Gali-Muhtasib. 2011. "A Journey Under the Sea: The Quest for Marine Anti-Cancer Alkaloids" Molecules 16, no. 11: 9665-9696. https://doi.org/10.3390/molecules16119665
APA StyleTohme, R., Darwiche, N., & Gali-Muhtasib, H. (2011). A Journey Under the Sea: The Quest for Marine Anti-Cancer Alkaloids. Molecules, 16(11), 9665-9696. https://doi.org/10.3390/molecules16119665