DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods

Abstract
:1. Peptides Presented on Phages
2. DNA Libraries

{kind=link}
{kind=link}
{kind=link}
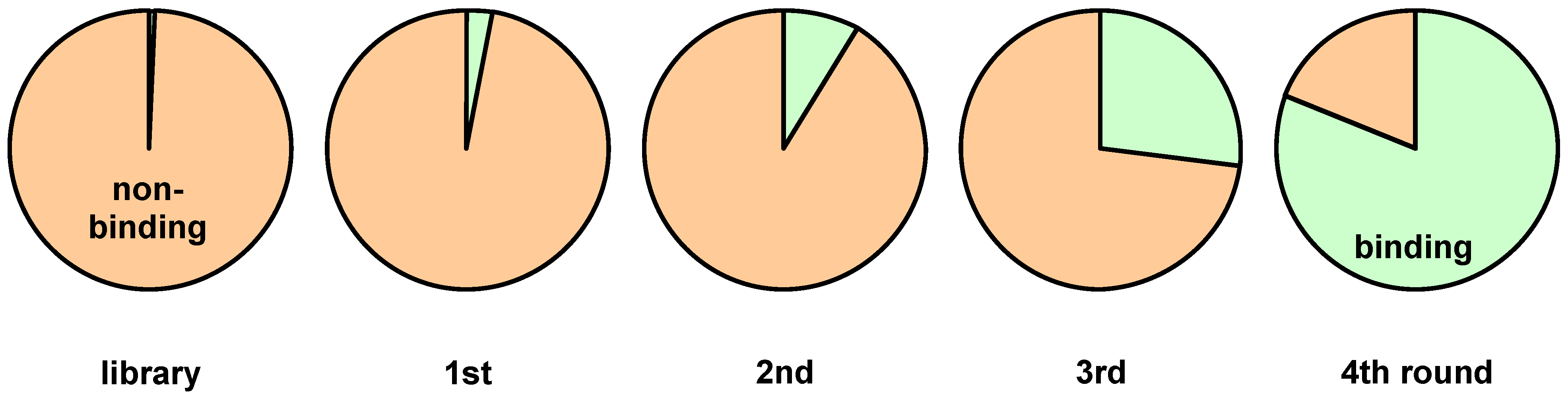{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| random positions | individual sequences | random positions | individual sequences |
|---|---|---|---|
| 7 | 1 × 109 | 12 | 4 × 1015 |
| 8 | 2 × 1010 | 13 | 1 × 1017 |
| 9 | 5 × 1011 | 14 | 2 × 1018 |
| 10 | 1 × 1013 | 15 | 3 × 1019 |
| 11 | 2 × 1014 | 16 | 6 × 1020 |
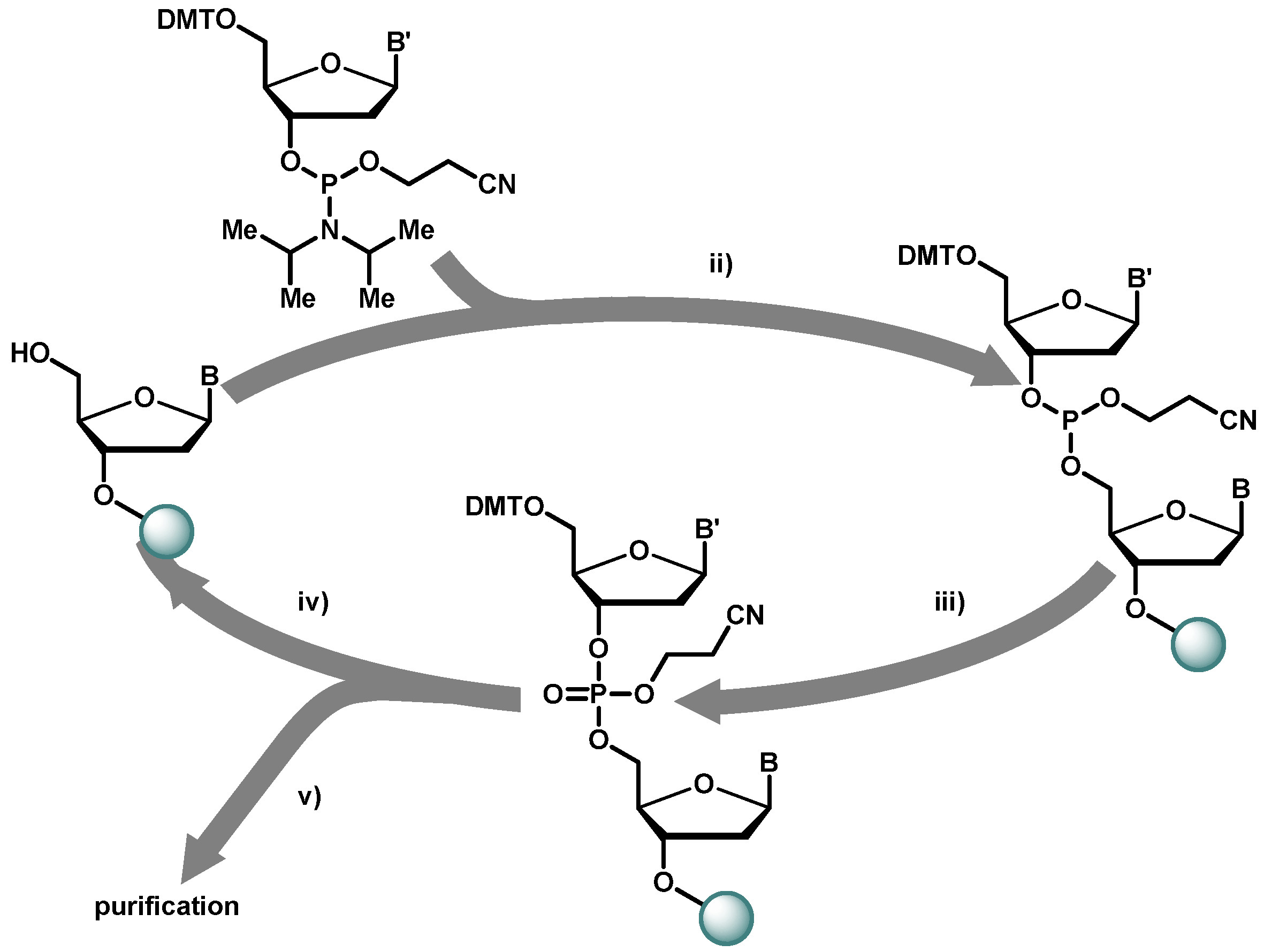
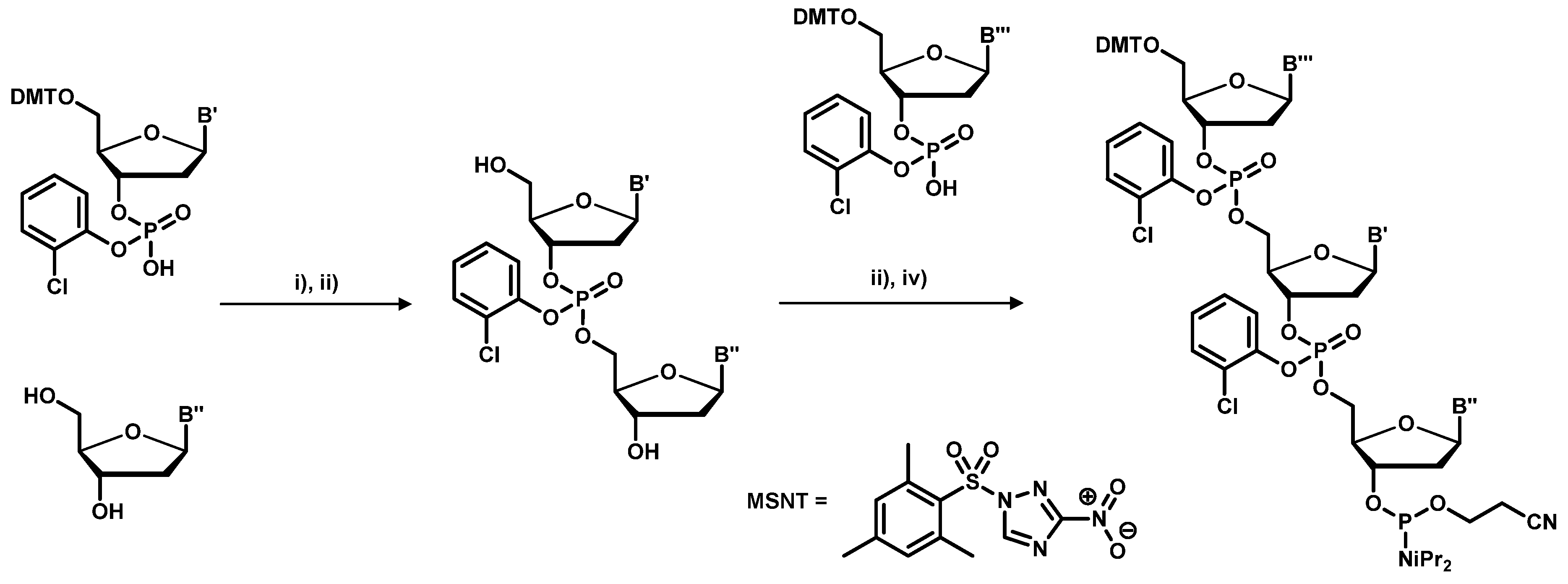
2.1. Chemical Synthesis of Library Inserts

2.2. Source of Variability
| TTT | Phe | TCT | Ser | TAT | Tyr | TGT | Cys |
| TTC | TCC | TAC | TGC | ||||
| TTA | Leu | TCA | TAA | STOP | TGA | STOP | |
| TTG | TCG | TAG | STOP | TGG | Trp | ||
| CTT | CCT | Pro | CAT | His | CGT | Arg | |
| CTG | CCC | CAC | CGC | ||||
| CTA | CCA | CAA | Gln | CGA | |||
| CTG | CCG | CAG | CGG | ||||
| ATT | Ile | ACT | Thr | AAT | Asn | AGT | Ser |
| ATG | ACG | AAC | AGC | ||||
| ATA | ACA | AAA | Lys | AGA | Arg | ||
| ATG | Met | ACG | AAG | AGG | |||
| GTT | Val | GCT | Ala | GAT | Asp | GGT | Gly |
| GTC | GCC | GAC | GGC | ||||
| GTA | GCA | GAA | Glu | GGA | |||
| GTG | GCG | GAG | GGG |
Improvement of statistic distribution by exclusion of rare codons
| G/T–A/C–T | Tyr, Ala, Asp, Ser | A/G–G/C–A | Thr, Arg, Gly, Ala |
| A/T–A/C–T | Tyr, Thr, Asn, Ser | G/C–A/G–C | His, Arg, Asp, Gly |
| C/T–A/C–T | Tyr, Pro, His, Ser | A/G–G/C–T | Gly, Ala, Thr, Ser |
| Ala | GCT | Arg | CGT | Asn | AAC | Asp | GAC | Cys | TGC |
| Gln | CAG | Glu | GAA | Gly | GGT | His | CAT | Ile | ATC |
| Leu | CTG | Lys | AAA | Met | ATG | Phe | TTC | Pro | CCG |
| Ser | TCT | Thr | ACT | Trp | TGG | Tyr | TAC | Val | GTT |

2.3. Biological Synthesis of Random Library Inserts
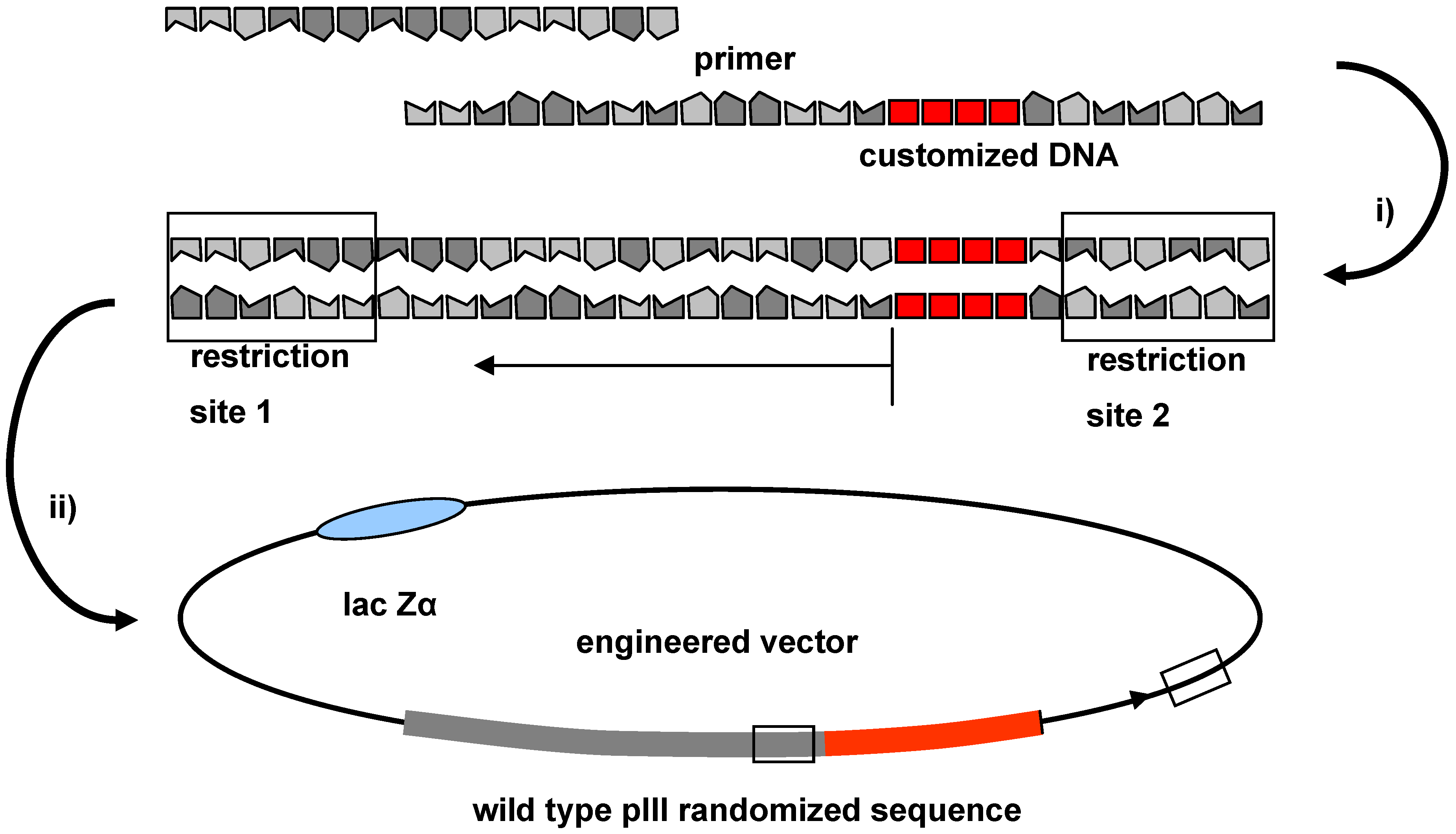
2.4. Cloning Technique for the Integration of Oligonucleotide Sequences into the Phage Genome

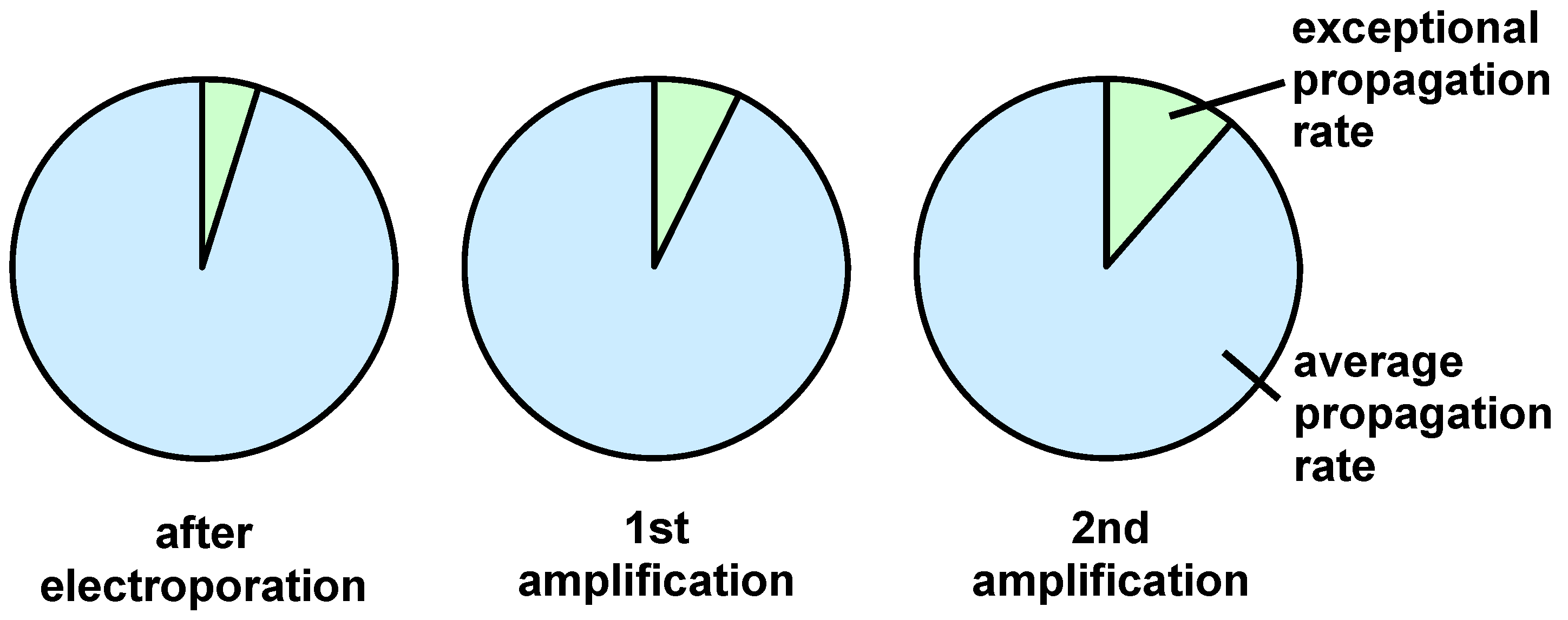
2.5. Determining the Variability of the Completed Peptide Library

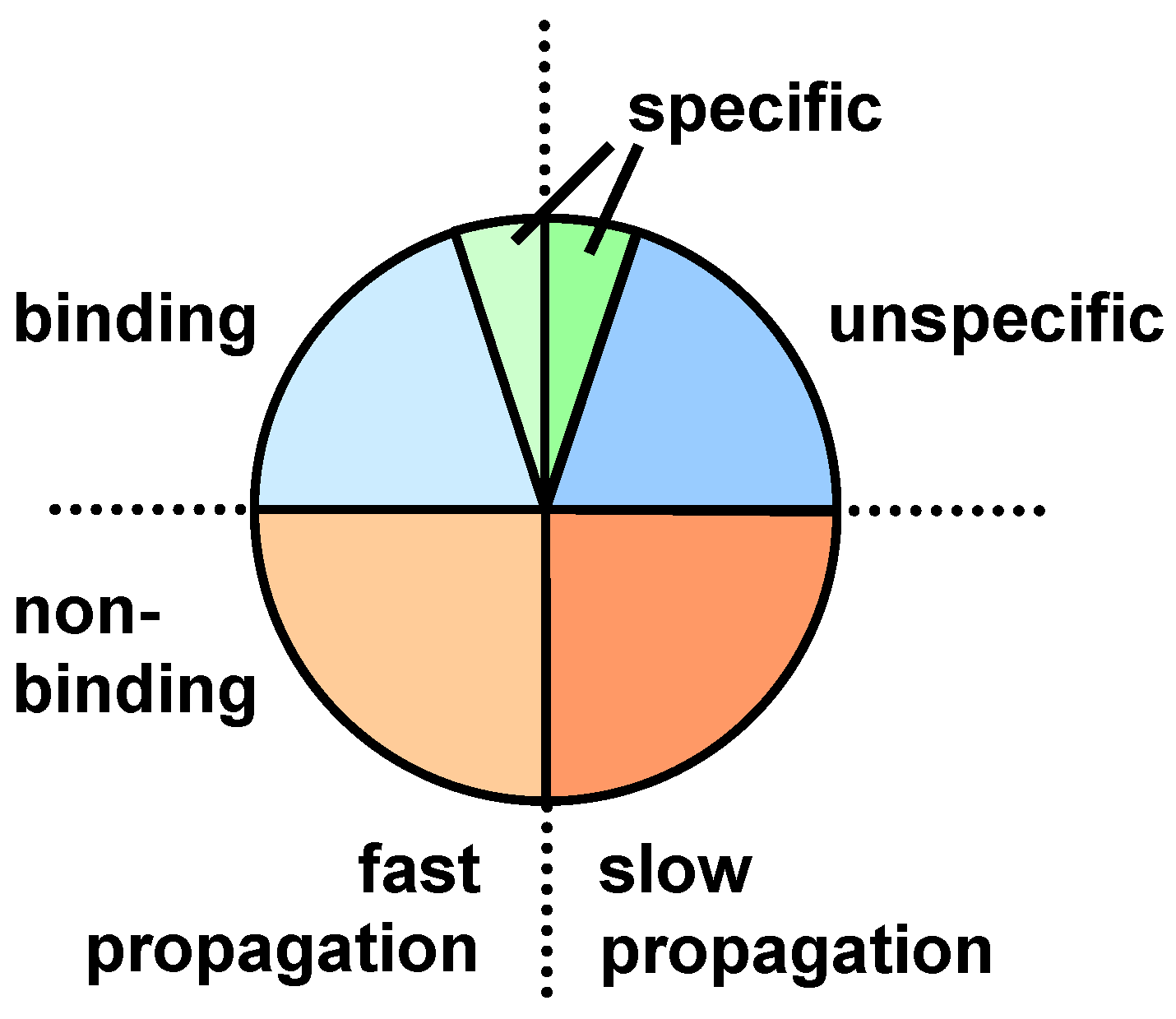
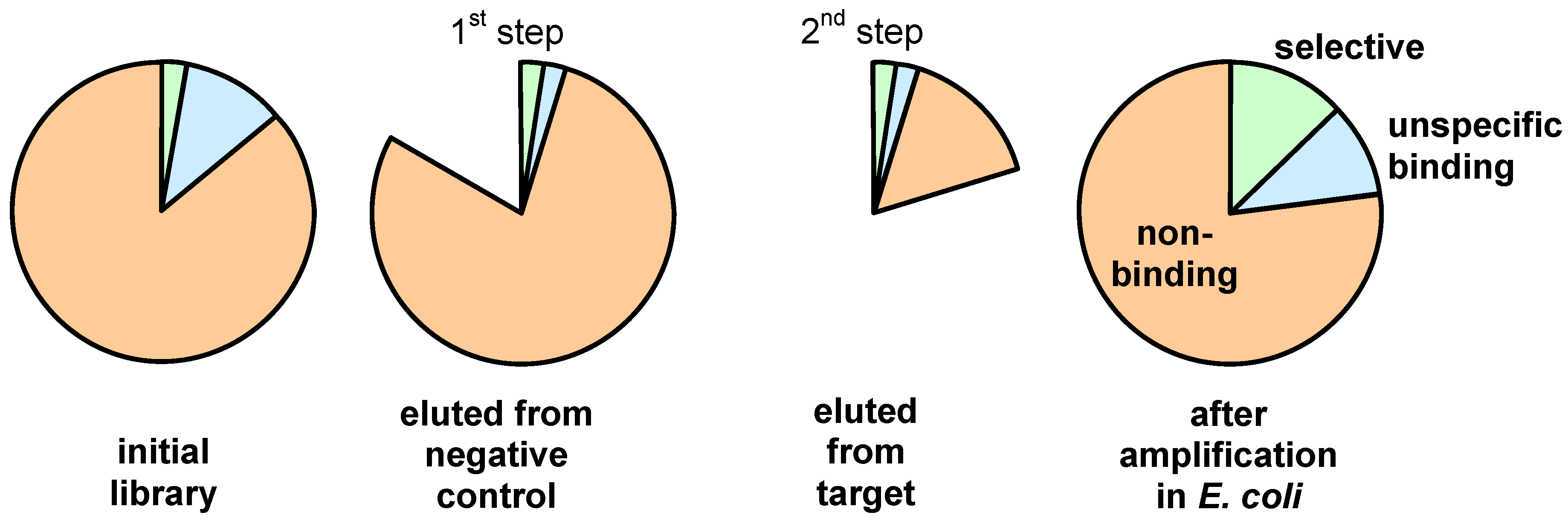
3. Screening Procedures




4. Sequence and Structural Motifs for Displayed Peptides
5. Conclusions
Acknowledgements
References
- Zoller, F.; Eisenhut, M.; Haberkorn, U.; Mier, W. Endoradiotherapy in cancer treatment-basic concepts and future trends. Eur. J. Pharmacol. 2009, 625, 55–62. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid-phase peptide synthesis. 3. An improved synthesis of bradykinin. Biochemistry 1964, 3, 1385–1390. [Google Scholar] [CrossRef]
- Merrifield, R.B.; Steward, J.M. Automated peptide synthesis. Nature 1965, 207, 522–523. [Google Scholar] [CrossRef]
- Chang, C.D.; Meienhofer, J. Solid-phase peptide synthesis using mild base cleavage of N-alpha-fluorenylmethoxycarbonylamino acids. Int. J. Pept. Protein Res. 1978, 11, 246–249. [Google Scholar]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef]
- Pang, W.; Tam, S.C.; Zheng, Y.T. Current peptide HIV type-1 fusion inhibitors. Antivir. Chem. Chemother. 2009, 20, 1–18. [Google Scholar] [CrossRef]
- Jackson, D.C.; Lau, Y.F.; Le, T.; Suhrbier, A.; Deliyannis, G.; Cheers, C.; Smith, C.; Zeng, W.; Brown, L.E. A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc. Natl. Acad. Sci. USA 2004, 101, 15440–15445. [Google Scholar]
- Graham, K.; Wang, Q.; Boy, R.G.; Eisenhut, M.; Haberkorn, U.; Mier, W. Synthesis and evaluation of intercalating somatostatin receptor binding peptide conjugates for endoradiotherapy. J. Pharm. Pharm. Sci. 2007, 10, 286. [Google Scholar]
- Reubi, J.C.; Schär, J.C.; Waser, B.; Wenger, S.; Heppeler, A.; Schmitt, J.S.; Mäcke, H.R. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur. J. Nucl. Med. 2000, 27, 273–282. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled cyclic RGD peptides as integrin αvβ3-targeted radiotracers: Maximizing binding affinity via bivalency. Bioconjug. Chem. 2009, 20, 2199–2213. [Google Scholar] [CrossRef]
- Kubas, H.; Schäfer, M.; Bauder-Wüst, U.; Eder, M.; Oltmanns, D.; Haberkorn, U.; Mier, W.; Eisenhut, M. Multivalent cyclic RGD ligands: influence of linker lengths on receptor binding. Nucl. Med. Biol. 2010, 37, 885–891. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Serizawa, T.; Sawada, T.; Matsuno, H.; Matsubara, T.; sato, T. A peptide motif recognizing a polymer stereoregularity. J. Am. Chem. Soc. 2005, 127, 13780–13781. [Google Scholar] [CrossRef]
- Chen, J.; Serizawa, T.; Komiyama, M. Peptides recognize photoresponsive targets. Angew. Chem. Int. Ed. 2009, 48, 2917–2920. [Google Scholar] [CrossRef]
- Seker, U.O.S.; Wilson, B.; Sahin, D.; Tamerler, C.; Sarikaya, M. Quantitaive affinity of genetically engineered repeating polypetides to inorganic surfaces. Biomacromolecules 2009, 10, 250–257. [Google Scholar] [CrossRef]
- Mao, C.; Liu, A.; Cao, B. Virus-based chemical and biological sensing. Angew. Chem. Int. Ed. Engl. 2009, 48, 6790–6810. [Google Scholar] [CrossRef]
- Haberkorn, U.; Eisenhut, M.; Altmann, A.; Mier, W. Endoradiotherapy with peptides-status and future development. Curr. Med. Chem. 2008, 15, 219–234. [Google Scholar] [CrossRef]
- Zang, L.; Shi, L.; Guo, J.; Pan, Q.; Wu, W.; Pan, X.; Wang, J. Screening and identification of a peptide specifically targeted to NCI-H1299 from a phage display peptide library. Cancer Lett. 2009, 281, 64–70. [Google Scholar] [CrossRef]
- He, M.; Taussig, M.J. Ribosome display: Cell-free protein display technology. Brief. Funct. Genomic Proteomic. 2002, 1, 204–212. [Google Scholar] [CrossRef]
- Kopsidas, G.; Carman, R.; Stutt, E.; Raicevic, A.; Roberts, A.; Siomos, M.A.; Dobric, N.; Pontes-Braz, L.; Coia, G. RNA mutagenesis yields highly diverse mRNA libraries for in vitro protein evolution. BMC Biotechnol. 2007, 7, 18. [Google Scholar] [CrossRef]
- Zahnd, C.; Amstutz, P.; Plückthun, A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 4, 269–279. [Google Scholar] [CrossRef]
- Georgiou, G.; Poetschke, H.L.; Stathopoulos, C.; Francisco, J.A. Practical applications of engineering gram-negative bacterial cell surfaces. Trends Biotechnol. 1993, 11, 6–10. [Google Scholar] [CrossRef]
- Little, M.; Fuchs, P.; Breitling, F.; Dubel, S. Bacterial surface presentation of proteins and peptides: an alternative to phage technology? Trends Biotechnol. 1993, 11, 3–5. [Google Scholar] [CrossRef]
- Shibasaki, S.; Maeda, H.; Ueda, M. Molecular display technology using yeast-arming technology. Anal. Sci. 2009, 25, 41–49. [Google Scholar] [CrossRef]
- Marvin, D.A. Filamentous phage structure, infection and assembly. Curr. Opin. Struct. Biol. 1998, 8, 150–158. [Google Scholar] [CrossRef]
- Krichevsky, A.; Rusnati, M.; Bugatti, A.; Waigmann, E.; Shohat, S.; Loyter, A. The fd phage and a peptide derived from its p8 coat protein interact with the HIV-1 Tat-NLS and inhibit its biological functions. Antivir. Res. 2005, 66, 67–78. [Google Scholar]
- Garufi, G.; Minenkova, O.; Lo Passo, C.; Pernice, I.; Felici, F. Display libraries on bacteriophage lambda capsid. Biotechnol. Ann. Rev. 2005, 11, 153–190. [Google Scholar] [CrossRef]
- Russel, M.; Model, P. Filamentous phage. In The Bacteriophages, 2nd; Calendar, R., Ed.; Oxford University Press Inc.: New York, NY, USA, 2005. [Google Scholar]
- Sidhu, S.S. Engineering M13 for phage display. Biomol. Eng. 2001, 18, 57–63. [Google Scholar] [CrossRef]
- Pande, J.; Szewczyk, M.M.; Grover, A.K. Phage display: Concept, innovations, applications and future. Biotechnol. Adv. 2010, 28, 849–858. [Google Scholar] [CrossRef]
- Krumpe, L.R.; Atkinson, A.J.; Smythers, G.W.; Kandel, A.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6, 4210–4222. [Google Scholar] [CrossRef]
- Cwirla, S.E.; Peters, E.A.; Berrett, R.W.; Dower, W.J. Peptides on phage: A vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. USA 1990, 87, 6378–6382. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef]
- Beaucage, S.L.; Caruthers, M.H. Deoxynucleoside phosphoramidites-A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981, 22, 1859–1862. [Google Scholar] [CrossRef]
- McBride, L.J.; Caruthers, M.H. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983, 24, 245–248. [Google Scholar] [CrossRef]
- Beaucage, S.L.; Iyer, R.P. Advances in the synthesis of oligonucleotides by the phosphoramidite approach. Tetrahedron 1992, 48, 2223–2311. [Google Scholar] [CrossRef]
- Bossi, L. Context effects: translation of UAG codon by suppressor tRNA is affected by the sequence following UAG in the message. J. Mol. Biol. 1983, 164, 73–87. [Google Scholar] [CrossRef]
- Pedersen, S. Escherichia coli ribosomes translate in vivo with variable rate. EMBO J. 1984, 3, 2895–2898. [Google Scholar]
- Grosjean, H.; Fiers, W. Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene 1982, 18, 199–209. [Google Scholar] [CrossRef]
- Makoff, A.J.; Oxer, M.D.; Romanos, M.A.; Fairweather, N.F.; Ballantine, S. Expression of tetanus toxin fragment C in E.coli: high level expression by removing rare codons. Nucleic Acids Res. 1989, 17, 10191–10202. [Google Scholar]
- Thanaraj, T.A.; Argos, P. Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 1996, 5, 1973–1983. [Google Scholar] [CrossRef]
- Phoenix, D.A.; Korotov, E. Evidence of rare codon clusters within Escherichia coli coding regions. FEMS Microbiol. Lett. 1997, 155, 63–66. [Google Scholar] [CrossRef]
- Widmann, M.; Clairo, M.; Dippon, J.; Pleiss, J. Analysis of the distribution of functionally relevant rare codons. BMC Genomics 2008, 9, 207. [Google Scholar] [CrossRef]
- Mena, M.A.; Daugherty, P.S. Automated design of degenerate codon libraries. Protein Eng. Des. Sel 2005, 18, 559–561. [Google Scholar] [CrossRef]
- Fellouse, F.A.; Wiesmann, C.; Sidhu, S.S. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc. Natl. Acad. Sci. USA 2004, 101, 12467–12472. [Google Scholar] [CrossRef]
- Belcourt, M.F.; Farabaugh, P.J. Ribosomal frameshifting in the yeast retrotransposon Ty: tRNAs induce slippage on a 7 nucleotide minimal site. Cell 1990, 62, 339–352. [Google Scholar] [CrossRef]
- Kayushin, A.L.; Korosteleva, M.D.; Miroshnikov, A.I.; Kosch, W.; Zubov, D.; Piel, N. A convenient approach to the synthesis of trinucleotide phosphoramidites-synthons for the generation of oligonucletide/peptide libraries. Nucleic Acids Res. 1996, 24, 1748–3755. [Google Scholar]
- Krumpe, L.R.H.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. Trinucleotide cassettes increase diversity of T7 phage-displayed peptide library. BMC Biotechol. 2007, 7, 65. [Google Scholar] [CrossRef]
- Virnekäs, B.; Ge, L.; Plückthun, A.; Schneider, K.C.; Wellnhofer, G.; Simon, E. Moroney Trinucleotide phosphoramidites: Ideal reagents for the synthesis of mixed oligonucleotides for random mutagenesis. Nucleic Acids Res. 1994, 22, 5600–5607. [Google Scholar]
- Yagodkin, A.; Azhayev, A.; Roivainen, J.; Antopolsky, M.; Kayushin, A.; Korosteleva, M.; Miroshnikov, A.; Randolph, J.; Mackie, A.H. Improved Synthesis of Trinucleotide Phosphoramidites and Generation of Randomized Oligonucleotide Libraries. Nucleos. Nucleot. Nucleic Acids 2007, 26, 473–497. [Google Scholar] [CrossRef]
- Gaytán, P.; Contreras-Zambrano, C.; Ortiz-Alvarado, M.; Morales-Pablos, A.; Yánez, J. TrimerDimer: an oligonucleotide-based saturation mutagenesis approach that removes redundant and stop codons. Nucleic Acids Res. 2009, 37, e125. [Google Scholar]
- Neuner, P.; Cortese, R.; Monaci, P. Codon-based mutagenesis using dimer-phosphoramidites. Nucleic Acids Res. 1998, 26, 1223–1227. [Google Scholar] [CrossRef]
- Stemmer, W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Smith, M. Synthetic DNA and biology (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1994, 33, 1214–1221. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Roberts, J.D.; Zakour, R.A. Rapid and efficient site-specific mutagenesis without phenotypic selection. Meth. Enzymol. 1987, 154, 367–382. [Google Scholar]
- Adams, T.M.; Schmoldt, H.U.; Kolmar, H. FACS screening of combinatorial peptide and protein librariers displayed on the surface of Escherichia coli cells. In Evolutionary Methods in Biotechnology: Clever Tricks for Directed Evolution; Brakmann, S., Schwienhorst, A., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004; Volume 1, pp. 31–46. [Google Scholar]
- DeGraaf, M.E.; Miceli, R.M.; Mott, J.E.; Fischer, H.D. Biochemical diversity in a phage display library of random decapeptides. Gene 1993, 128, 13–17. [Google Scholar] [CrossRef]
- Rodi, D.J.; Soares, A.S.; Makowski, L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J. Mol. Biol. 2002, 322, 1039–1052. [Google Scholar] [CrossRef]
- Makowski, L.; Soares, A. Estimating the diversity of peptide populations from limited sequence data. Bioinformatics 2003, 19, 183–489. [Google Scholar] [CrossRef]
- Mandava, S.; Makowski, L.; Devarapalli, S.; Uzubell, J.; Rodi, D.J. RELIC-A bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics 2004, 4, 1439–1460. [Google Scholar] [CrossRef]
- Dias-Neto, E.; Nunes, D.N.; Giordano, R.J.; Sun, J.; Botz, G.H.; Yang, K.; Setubal, J.C.; Pasqualini, R.; Arap, W. Next-generation phage display: integrating and comparing available molecular tools to enable cost-effective high-throughput analysis. PLoS ONE 2009, 4, e8338. [Google Scholar]
- Noren, K.A.; Noren, C.J. Construction of high-complexity combinatorial phage display peptide libraries. Methods 2001, 23, 169–178. [Google Scholar] [CrossRef]
- Bratkovic, T. Progress in phage display: Evolution of the technique and its applications. Cell. Mol. Life Sci. 2010, 67, 749–767. [Google Scholar] [CrossRef]
- Sun, X.; Niu, G.; Yan, Y.; Yang, M.; Chen, K.; Ma, Y.; Chan, N.; Shen, B.; Chen, X. Phage display-derived Peptides for osteosarcoma imaging. Clin. Cancer Res. 2010, 16, 4268–4277. [Google Scholar] [CrossRef]
- Rajotte, D.; Arap, W.; Hagedorn, M.; Koivunen, E.; Pasqualini, R.; Ruoslathi, E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J. Clin. Invest. 1998, 102, 430–437. [Google Scholar] [CrossRef]
- Fleming, T.J.; Sachdeva, M.; Delic, M.; Beltzer, J.; Wescott, C.R.; Devlin, M.; Lander, R.C.; Nixon, A.E.; Roschke, V.; Hilbert, D.M.; Sexton, D.J. Discovery of high-affinity peptide binders to BLyS by phage display. J. Mol. Recognit. 2005, 18, 94–102. [Google Scholar] [CrossRef]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar]
- Binz, H.K.; Amstutz, P.; Plückthun, A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005, 23, 1257–1268. [Google Scholar] [CrossRef]
- Skerra, A. Alternative non-antibody scaffolds for molecular recognition. Curr. Opin. Biotechnol. 2007, 18, 295–304. [Google Scholar] [CrossRef]
- Kolmar, H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef]
- McConnell, S.J.; Kendall, M.L.; Reilly, T.M.; Hoess, R.H. Constrained peptide libraries as a tool for finding mimotopes. Gene 1994, 151, 115–118. [Google Scholar] [CrossRef]
- Pini, A.; Bracci, L. Phage display of antibody fragments. Curr. Protein Pept. Sci. 2000, 1, 155–169. [Google Scholar] [CrossRef]
- Filpula, D. Antibody engineering and modification technologies. Biomol. Eng. 2007, 24, 201–215. [Google Scholar] [CrossRef]
- Ladner, R.C. Constrained peptides as binding entities. Trends Biotechnol. 1995, 13, 426–430. [Google Scholar] [CrossRef]
- McLafferty, M.A.; Kent, R.B.; Ladner, R.C.; Markland, W. M13 bacteriophage displaying disulfide-constrained microproteins. Gene 1993, 128, 29–36. [Google Scholar] [CrossRef]
- Uchiyama, F.; Tanaka, Y.; Minari, Y.; Tokui, N. Designing scaffolds of peptides for phage display libraries. J. Biosci. Bioengineer. 2005, 99, 448–456. [Google Scholar] [CrossRef]
- Heinis, C.; Rutherford, T.; Freund, S.; Winter, G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol. 2009, 5, 502–507. [Google Scholar] [CrossRef]
- Moffet, D.A.; Hecht, M.H. De novo proteins from combinatorial libraries. Chem. Rev. 2001, 101, 3191–3203. [Google Scholar] [CrossRef]
- Tian, F.; Tsao, M.L.; Schultz, P.G. A phage display system with unnatural amino acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. [Google Scholar] [CrossRef]
- Liu, C.C.; Mack, A.V.; Tsao, M.L.; Mills, J.H.; Lee, H.S.; Choe, H.; Farzan, M.; Schultz, P.G.; Smider, V.V. Protein evolution with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2008, 105, 17688–17693. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lindner, T.; Kolmar, H.; Haberkorn, U.; Mier, W. DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods. Molecules 2011, 16, 1625-1641. https://doi.org/10.3390/molecules16021625
Lindner T, Kolmar H, Haberkorn U, Mier W. DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods. Molecules. 2011; 16(2):1625-1641. https://doi.org/10.3390/molecules16021625
Chicago/Turabian StyleLindner, Thomas, Harald Kolmar, Uwe Haberkorn, and Walter Mier. 2011. "DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods" Molecules 16, no. 2: 1625-1641. https://doi.org/10.3390/molecules16021625
APA StyleLindner, T., Kolmar, H., Haberkorn, U., & Mier, W. (2011). DNA Libraries for the Construction of Phage Libraries: Statistical and Structural Requirements and Synthetic Methods. Molecules, 16(2), 1625-1641. https://doi.org/10.3390/molecules16021625