From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins



Abstract
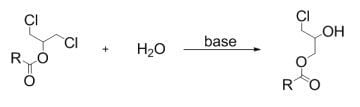
:
1. Introduction
2. Results and Discussion
3. Experimental
3.1. General
3.2. Procedure for the syntheses of 2-chloro-1-(chloromethyl)ethyl esters 1
3.3. Procedure for the syntheses of 3-chloro-2-hydroxy-1-propyl esters 2
3.4. Procedure for the syntheses of 3-chloro-2-hydroxy-1-propyl esters 2a and 2d using different inorganic bases
3.5. Procedure for the multigram-scale syntheses of 3-chloro-2-hydroxy-1-propyl 2,2-dimethylpropanoate (2b)
4. Conclusions
Acknowledgements
References and Notes
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Uses of a versatile Raw Material; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Zhou, C.H.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Eilting, J.; Irawadi, K.; Leschinski, J.; Lindner, F. Improved utilisation of renewable resources: New important derivatives of glycerol. Green Chem. 2008, 10, 13–30. [Google Scholar] [CrossRef]
- Johnson, D.T.; Taconi, K.A. The glycerin glut: Options for the value-added conversion of crude glycerol resulting from biodiesel production. Environ. Prog. 2007, 26, 338–348. [Google Scholar] [CrossRef]
- Meher, L.C.; Vidya Sagar, D.; Naik, S.N. Technical aspects of biodiesel production by transesterification--a review. Renew. Sustain. Energy Rev. 2006, 10, 248–268. [Google Scholar] [CrossRef]
- Janaun, J.; Ellis, N. Perspectives on biodiesel as a sustainable fuel. Renew. Sustain. Energy Rev. 2010, 14, 1312–1320. [Google Scholar] [CrossRef]
- Chiu, C.-W.; Goff, M.J.; Suppes, G.J. Distribution of methanol and catalysts between biodiesel and glycerin phases. AIChE J. 2005, 51, 1274–1278. [Google Scholar] [CrossRef]
- Yori, J.C.; D’Ippolito, S.A.; Pieck, C.L.; Vera, C.R. Deglycerolization of Biodiesel Streams by Adsorption Over Silica Beds. Energ. Fuel. 2006, 21, 347–353. [Google Scholar] [CrossRef]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Della Pina, C. From Glycerol to Value-Added Products. Angew. Chem. Inter. Ed. 2007, 46, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, M.; Ciriminna, R.; Kimura, H.; Rossi, M.; Pina, C.D. Recent advances in the conversion of bioglycerol into value-added products. Eur. J. Lipid Sci. Technol. 2009, 111, 788–799. [Google Scholar] [CrossRef]
- Villorbina, G.; Tomàs, A.; Escribà, M.; Oromí-Farrús, M.; Eras, J.; Balcells, M.; Canela, R. Combining AlCl3·6H2O and an ionic liquid to prepare chlorohydrin esters from glycerol. Tetrahedron Lett. 2009, 50, 2828–2830. [Google Scholar] [CrossRef]
- Escribà, M.; Eras, J.; Duran, M.; Simon, S.; Butchosa, C.; Villorbina, G.; Balcells, M.; Canela, R. From glycerol to chlorohydrin esters using a solvent-free system. Microwave irradiation versus conventional heating. Tetrahedron 2009, 65, 10370–10376. [Google Scholar] [CrossRef]
- Eras, J.; Balcells, M.; Canela, R. Glycerol as a starting material to prepare palmitate derivatives. Afinidad 2007, 64, 203–206. [Google Scholar]
- Beger, J.; Jacobi, R.; Rehbeil, U.; Knoll, E. Mehrfunktionelle N-Tenside. XIV: Synthesen und kritische Mizellbildungskonzentrationen von quartären Ammoniumsalzen mit Carbonsäurederivatfunktionen (Multifunctional N-surfactants. XIV: Synthesis and micellar critical concentration of quaternary ammonium salts with carboxylic acid derivatives functions). Tenside, Surfactants, Deterg. 1992, 29, 328–332. [Google Scholar]
- Hamaguchi, S.; Ohashi, T.; Watanabe, K. Lipase-catalyzed stereoselective hydrolysis of 2-acyloxy-3-chloropropyl-p-toluenesulfonate. Agric. Biol. Chem. 1986, 50, 375–380. [Google Scholar]
- Junk, T.; Pappalardo, G.C.; Irgolic, K.J. Synthesis and characterization of rac-1,2-bis(palmitoyloxy)-3-propyl (2-trimethylarsonioethyl)phosphonate, an arsenic-containing phosphonolipid. Appl. Organometal. Chem. 1990, 4, 103–109. [Google Scholar] [CrossRef]
- Kapoor, M.; Anand, N.; Ahmad, K.; Koul, S.; Chimni, S.S.; Taneja, S.C.; Qazi, G.N. Synthesis of β-adrenergic blockers (R)-(-)-nifenalol and (S)-(+)-sotalol via a highly efficient resolution of a bromohydrin precursor. Tetrahedron Asymmetry 2005, 16, 717–725. [Google Scholar] [CrossRef]
- Kolb, H.C.; Bennani, Y.L.; Sharpless, K.B. Short and practical synthess of (R)-(-)-carnitine and (R)-(-)-γ-amino-β-hydroxybutyric acid (GABOB). Tetrahedron Asymmetry 1993, 4, 133–141. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. A simplified procedure for the stereospecific transformation of 1,2-diols into epoxides. Tetrahedron 1992, 48, 10515–10530. [Google Scholar] [CrossRef]
- Marzi, M.; Minetti, P.; Moretti, G.; Tinti, M.O.; De Angelis, F. Efficient enantioselective synthesis of (R)-(-)-carnitine from glycerol. J. Org. Chem. 2000, 65, 6766–6769. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, K. Positive-charging electrophotographic toner using bisphenol A-free linear polyester binder. JP2005326606, 24 November 2005. [Google Scholar]
- Moschidis, M.C. Synthesis of 1,2-diacyloxypropyl-3-(1′,2′-diacyl-sn-glycero)-phosphonate. Chem. Phys. Lipids 1985, 36, 297–302. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Papahatjis, D.P.; Claremon, D.A.; Magolda, R.L.; Dolle, R.E. Total synthesis of ionophore antibiotic X-14547A. J. Org. Chem. 1985, 50, 1440–1456. [Google Scholar] [CrossRef]
- Schmitt, J.D.; Nixon, A.B.; Emilsson, A.; Daniel, L.W.; Wykle, R.L. A facile synthesis of 1-O-alkyl-2-(R)-hydroxypropane-3-phosphonocholine (lyso-phosphono-platelet activating factor). Chem. Phys. Lipids 1992, 62, 263–268. [Google Scholar] [CrossRef]
- Watson, H.G.; Fung, Y.M.; Gredley, M.; Bird, G.J.; Jackson, W.R.; Gountzos, H.; Matthews, B.R. Asymmetric syntheses of (+)-diltiazem hydrochloride. J. Chem. Soc. Ser. Chem. Commun. 1990, 1018–1019. [Google Scholar] [CrossRef]
- Wilen, S.H.; Delguzzo, L.; Saferstein, R. Experimental evidence for AcO-7 neighboring group participation. Tetrahedron 1987, 43, 5089–5094. [Google Scholar] [CrossRef]
- Williams, J.R.; Boehm, J.C. The syntheses of 3[beta]-steroidal diacylglyceryl sulfides, sulfoxides, and sulfones. Steroids 1995, 60, 321–323. [Google Scholar] [CrossRef]
- Leggetter, B.E.; Brown, R.K. The influence of substituents on the ease and direction of ring opening in the LiAlH4-AlCl3 reductive cleavage of substituted 1,3-dioxolanes. Can. J. Chem. 1964, 42, 990–1004. [Google Scholar] [CrossRef]
- Leggetter, B.E.; Brown, R.K. Anomalous ring cleavage during lithium aluminum hydride - aluminum chloride reduction of 2,2,4,4-tetraalkylated-1,3-dioxolanes. Can. J. Chem. 1964, 42, 1005–1008. [Google Scholar] [CrossRef]
- Eras, J.; Mendez, J.J.; Balcells, M.; Canela, R. Chlorotrimethylsilane: A Suitable Reagent for the Synthesis of Chlorohydrin Esters. J. Org. Chem. 2002, 67, 8631–8634. [Google Scholar] [CrossRef] [PubMed]
- Mormann, W.; Demeter, J.; Wagner, T. Selectivity in the acylation of partially silylated hydroxy polymers - a study with trimethylsilyl cellulose and model compounds. Acta Polym. 1999, 50, 20–27. [Google Scholar] [CrossRef]
Sample Availability: Samples are available from the authors. |


{kind=link}
{kind=link}

| Entry | R | Solvent | Base | Yielda of 2 (%) |
|---|---|---|---|---|
| a | CH3(CH2)14– | 1,4-Dioxane | Sodium carbonate | 48 |
| a | CH3(CH2)14– | 1,4-Dioxane | 1-Butylimidazole | 49 |
| a | CH3(CH2)14– | 1-Butanol | Sodium carbonate | – |
| a | CH3(CH2)14– | 1-Butanol | 1-Butylimidazole | – |
| b | (CH3)3C– | 1,4-Dioxane | Sodium carbonate | 62 |
| b | (CH3)3C– | 1,4-Dioxane | 1-Butylimidazole | 61 |
| b | (CH3)3C– | 1-Butanol | Sodium carbonate | 62 |
| b | (CH3)3C– | 1-Butanol | 1-Butylimidazole | 45 |
| c | CH3CH2(CH3)2C– | 1,4-Dioxane | Sodium carbonate | 36 |
| c | CH3CH2(CH3)2C– | 1,4-Dioxane | 1-Butylimidazole | 30 |
| c | CH3CH2(CH3)2C– | 1-Butanol | Sodium carbonate | – |
| c | CH3CH2(CH3)2C– | 1-Butanol | 1-Butylimidazole | 23 |
| d | Ph(CH3)2C– | 1,4-Dioxane | Sodium carbonate | 35 |
| d | Ph(CH3)2C– | 1,4-Dioxane | 1-Butylimidazole | 33 |
| d | Ph(CH3)2C– | 1-Butanol | Sodium carbonate | 7 |
| d | Ph(CH3)2C– | 1-Butanol | 1-Butylimidazole | 33 |

| Entry | R | Base | Yielda of 2 (%) |
|---|---|---|---|
| a | CH3(CH2)14– | Sodium carbonate | 48 |
| a | CH3(CH2)14– | Potassium carbonate | 25 |
| a | CH3(CH2)14– | Lithium carbonate | 35 |
| d | Ph(CH3)2C– | Sodium carbonate | 35 |
| d | Ph(CH3)2C– | Potassium carbonate | 23 |
| d | Ph(CH3)2C– | Lithium carbonate | 27 |
© 2011 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Solarte, C.; Escribà, M.; Eras, J.; Villorbina, G.; Canela, R.; Balcells, M. From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins. Molecules 2011, 16, 2065-2074. https://doi.org/10.3390/molecules16032065
Solarte C, Escribà M, Eras J, Villorbina G, Canela R, Balcells M. From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins. Molecules. 2011; 16(3):2065-2074. https://doi.org/10.3390/molecules16032065
Chicago/Turabian StyleSolarte, Carmen, Marc Escribà, Jordi Eras, Gemma Villorbina, Ramon Canela, and Mercè Balcells. 2011. "From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins" Molecules 16, no. 3: 2065-2074. https://doi.org/10.3390/molecules16032065
APA StyleSolarte, C., Escribà, M., Eras, J., Villorbina, G., Canela, R., & Balcells, M. (2011). From Symmetric Glycerol Derivatives to Dissymmetric Chlorohydrins. Molecules, 16(3), 2065-2074. https://doi.org/10.3390/molecules16032065