Molecular Docking of Aromatase Inhibitors

Abstract
:1. Introduction


2. Results and Discussion
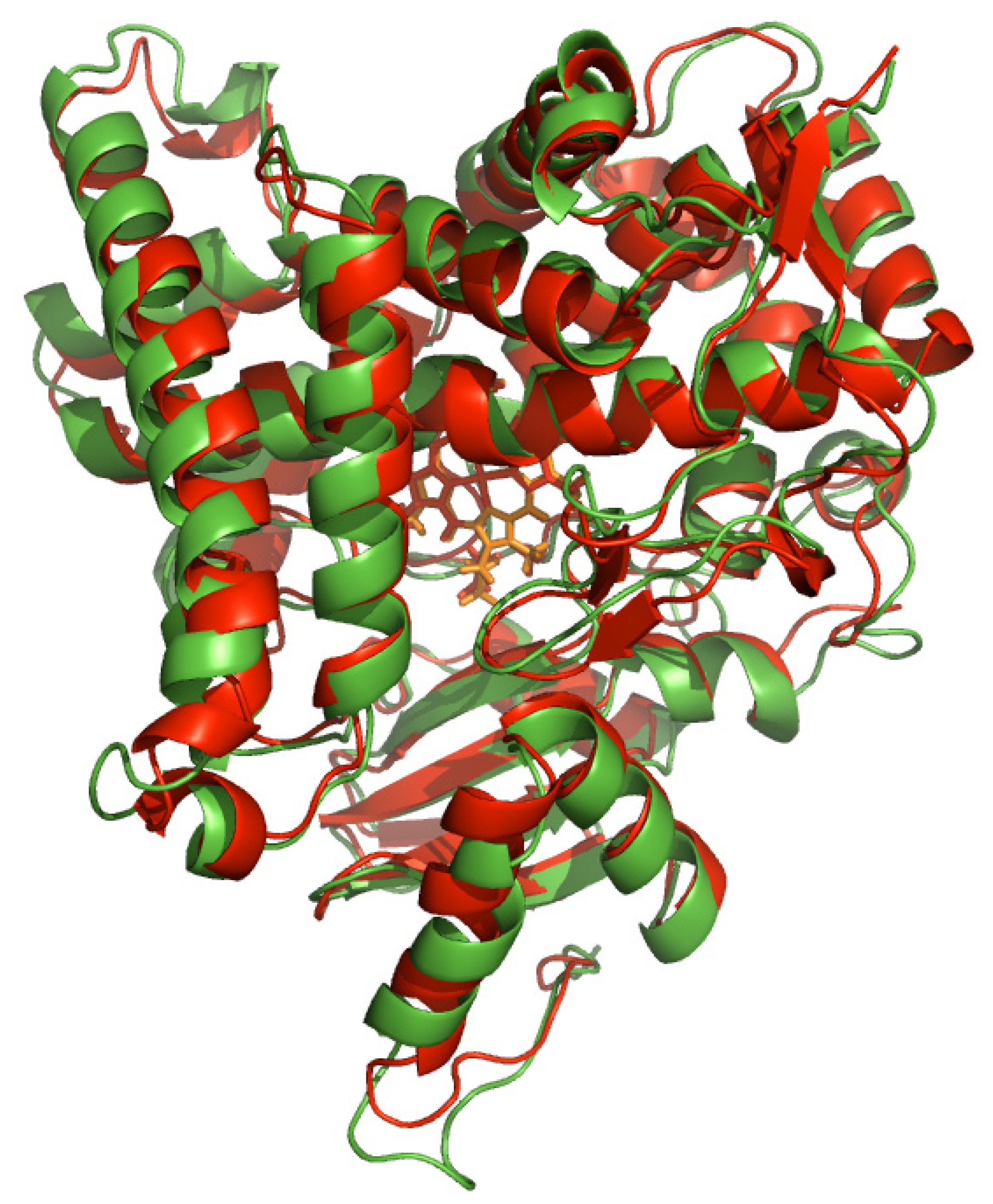
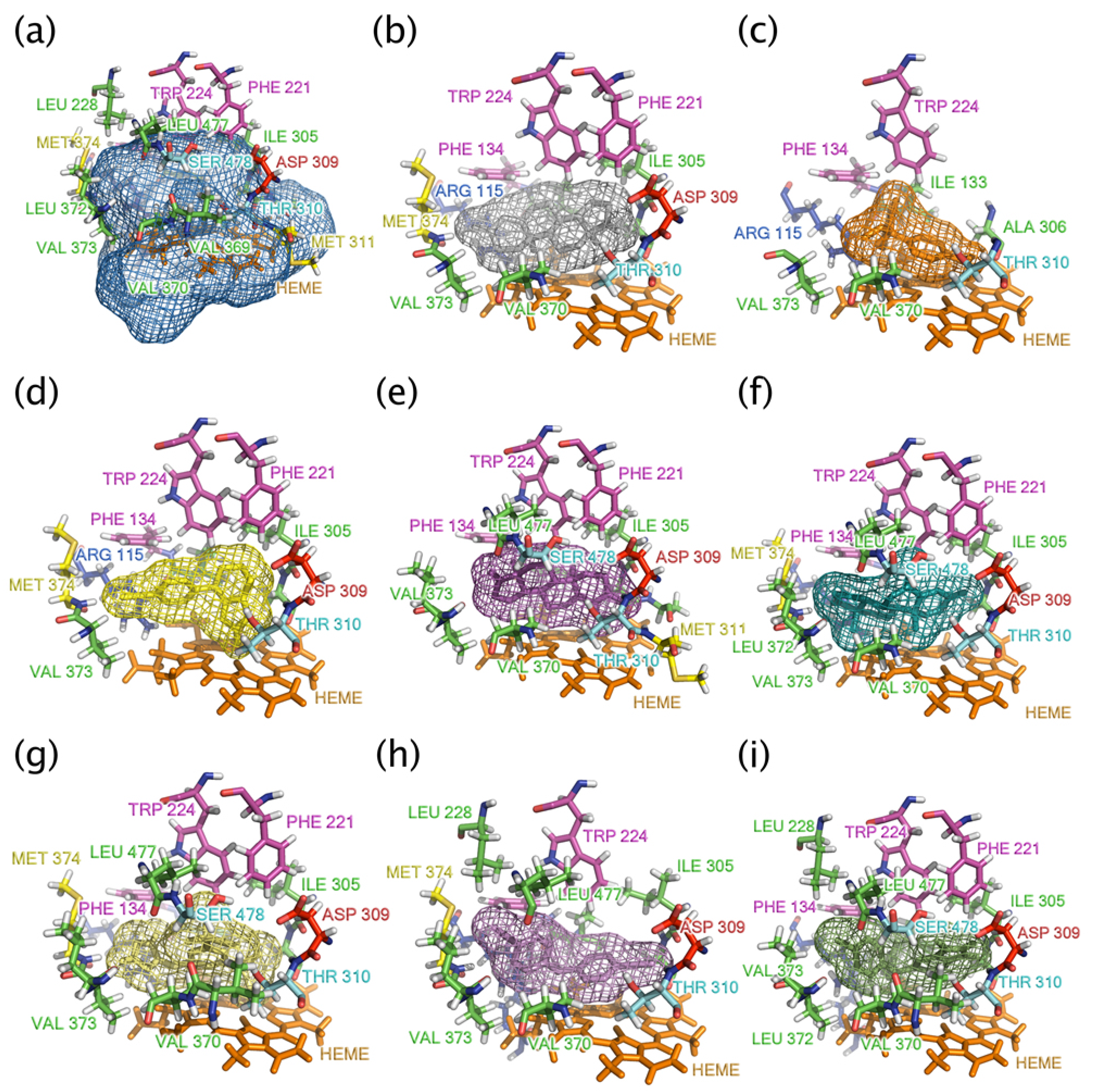
2.1. Analyzing the Binding Site of Aromatase
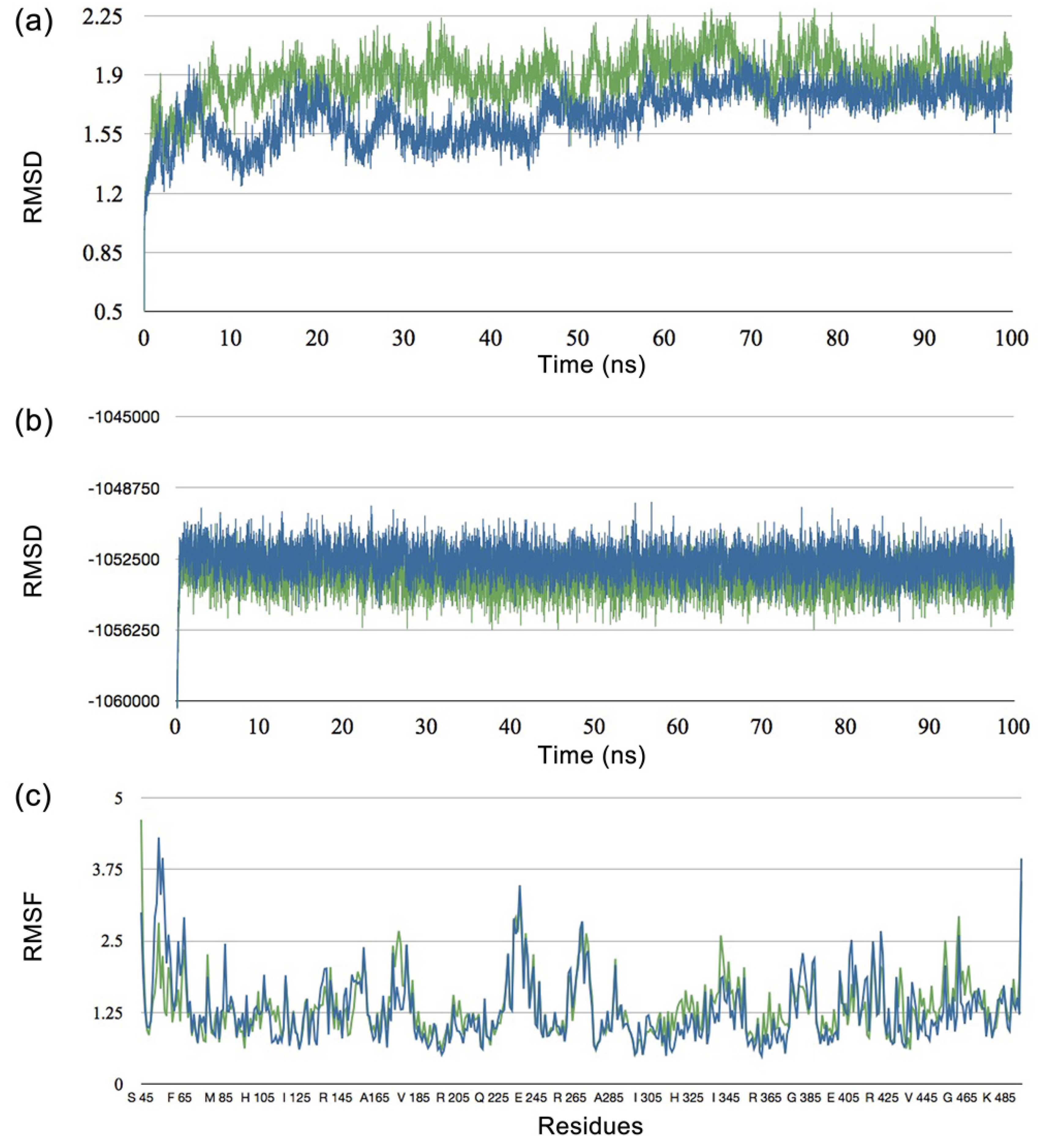
2.2. Molecular Dynamics Studies


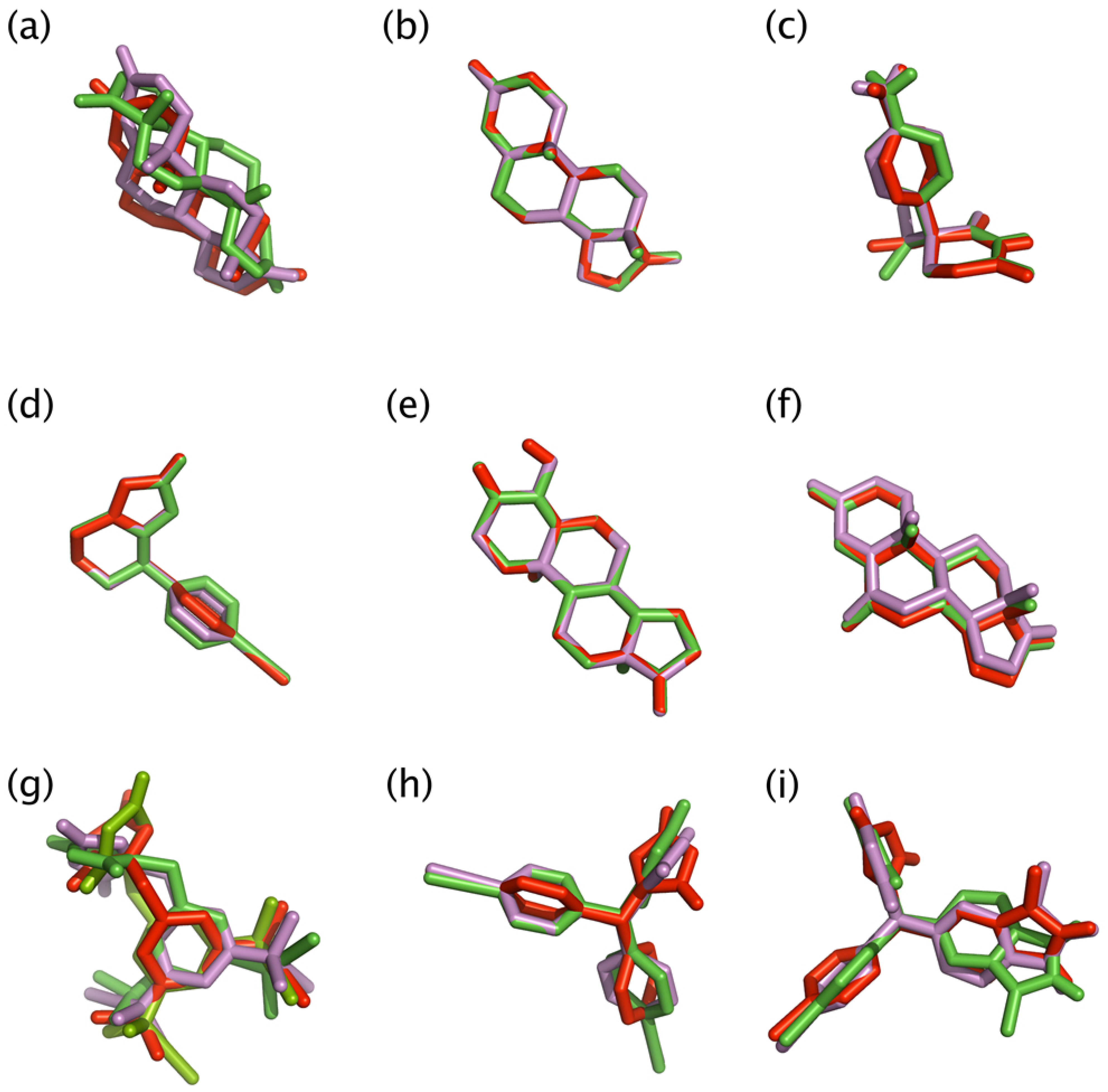
2.3. Docking Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Number of rotational bonds | Binding Energy(ΔGb) (kcal/mol) | Inhibition constant(Ki) (nM) | IC50 (nM) | |
|---|---|---|---|---|---|
| Cluster | Best Energy | ||||
| Androstenedione a | 0 | 100 | −11.04 | 8.06 | |
| Aminoglutethimide b | 3 | 71 | −7.85 | 1750.00 | 20000 |
| 3 | 23 | −7.65 | 2480.00 | 20000 | |
| Fadrozole c | 1 | 61 | −7.95 | 1490.00 | 30 |
| 1 | 39 | −8.18 | 1010.00 | 30 | |
| Formestane c | 1 | 100 | −10.99 | 8.77 | 30 |
| Exemestane c | 0 | 100 | −11.30 | 5.24 | 15 |
| Anastrozole d | |||||
| major cluster | 4 | 45 | −9.09 | 218.04 | 8 |
| minor cluster | 4 | 38 | −9.32 | 147.55 | 8 |
| Letrozole d | 3 | 99 | −8.78 | 367.20 | 2 |
| Vorozole d | 3 | 83 | −8.64 | 462.16 | |
| 3 | 17 | −8.66 | 451.26 | ||
| Estrone e | 1 | 100 | −10.30 | 28.39 | |
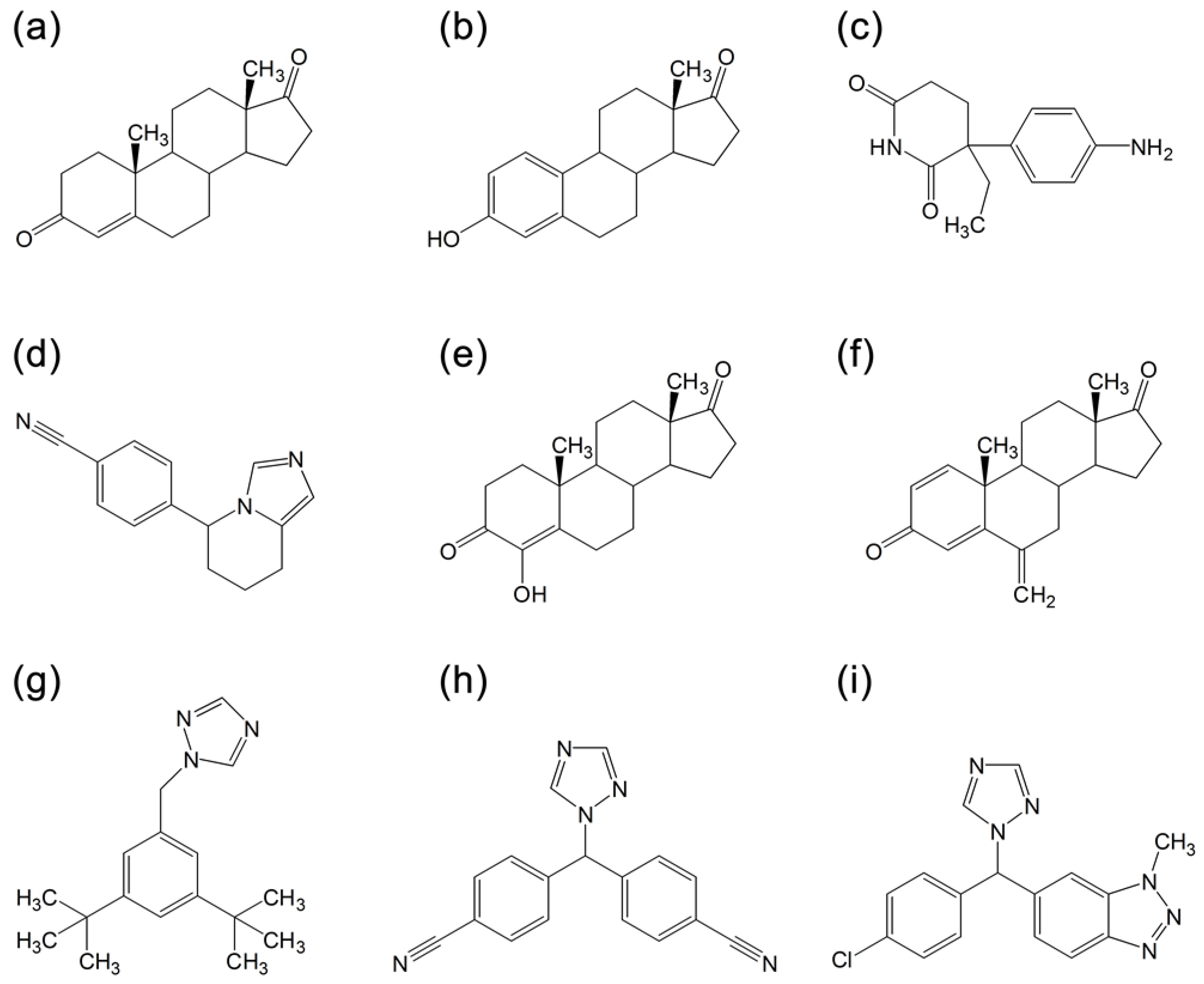
2.4. Analyzing the Docking Results of Aromatase Inhibitors
| Qm | Energy (kcal/mol) | μ(debye) | HOMO(eV) | LUMO(eV) | HOMO-LUMOgap(eV) | RMSD | Log P | |
|---|---|---|---|---|---|---|---|---|
| Androstenedione | 0.211 | -890.133 | 3.746 | −0.232 | −0.046 | 0.186 | 0 | 2.7 |
| Aminoglutethimide | 0.280 | -765.007 | 3.626 | −0.206 | −0.025 | 0.181 | 1.04 | 1.2 |
| Fadrozole | 0.202 | -705.565 | 5.687 | −0.218 | −0.061 | 0.157 | 0.16 | 2.1 |
| Formestane | 0.224 | -965.357 | 2.178 | −0.223 | −0.053 | 0.169 | 0.08 | 2.6 |
| Exemestane | 0.206 | -926.993 | 4.430 | −0.235 | −0.062 | 0.173 | 0 | 3.1 |
| Anastrozole | 0.232 | -932.972 | 4.982 | −0.265 | −0.036 | 0.229 | 1.43 | 2.1 |
| Letrozole | 0.201 | -928.146 | 3.911 | −0.269 | −0.073 | 0.196 | 1 | 2.7 |
| Vorozole | 0.200 | -1446.710 | 1.871 | −0.244 | −0.051 | 0.193 | 0.26 | 3.1 |
| Estrone | 0.212 | -849.592 | 3.981 | −0.210 | −0.015 | 0.195 | 0 | 3.1 |
| Ligand | H-bond Interaction | Metal coordination | Hydrophobic Interaction |
|---|---|---|---|
| Androstenedione | M374, T310 | R115, F134, F221, W224, I305, A306, D309, | |
| V370, V373, M374 | |||
| Aminoglutethimide | A306, T310 | R115, I133, F134, W224, V370, V373 | |
| Fadrozole | Azole-Heme | R115, F134, F221, W224, I305, A306, D309, | |
| T310, V373, M374 | |||
| Formestane | A306, T310 | I133, F134, F221, W224, I305, A307, D309, | |
| M311, V370, L372, V373, S478 | |||
| Exemestane | R115, M374 | F134, F221, W224, A306, D309, T310, | |
| V370, L372, V373, L477, S478 | |||
| Anastrozole | |||
| major cluster | L372 | Azole-Heme | R115, I133, F134, F221, W224, I305, A306,D309, T310, V370, V373, M374, L477, S478 |
| minor cluster | Azole-Heme | R115, I133, F134, F221, W224, I305, A306, D309, T310, V369, V370, R435, L477, S478 | |
| Letrozole | R115, I133, F134, W224, L228, I305, A306, D309, T310, V370, L372, V373, M374, R435, L477 | ||
| Vorozole | I305, D309 | Azole-Heme | R115, I133, F134, F221, W224, L228, A306, |
| T310, V369, V370, L372, V373, R435, L477, S478 | |||
| Estrone | M374 | R115, F134, A306, T310, V370, L372, V373 |

2.5. Key Interaction Residues
2.6. Analysis of Structure-activity Relationship
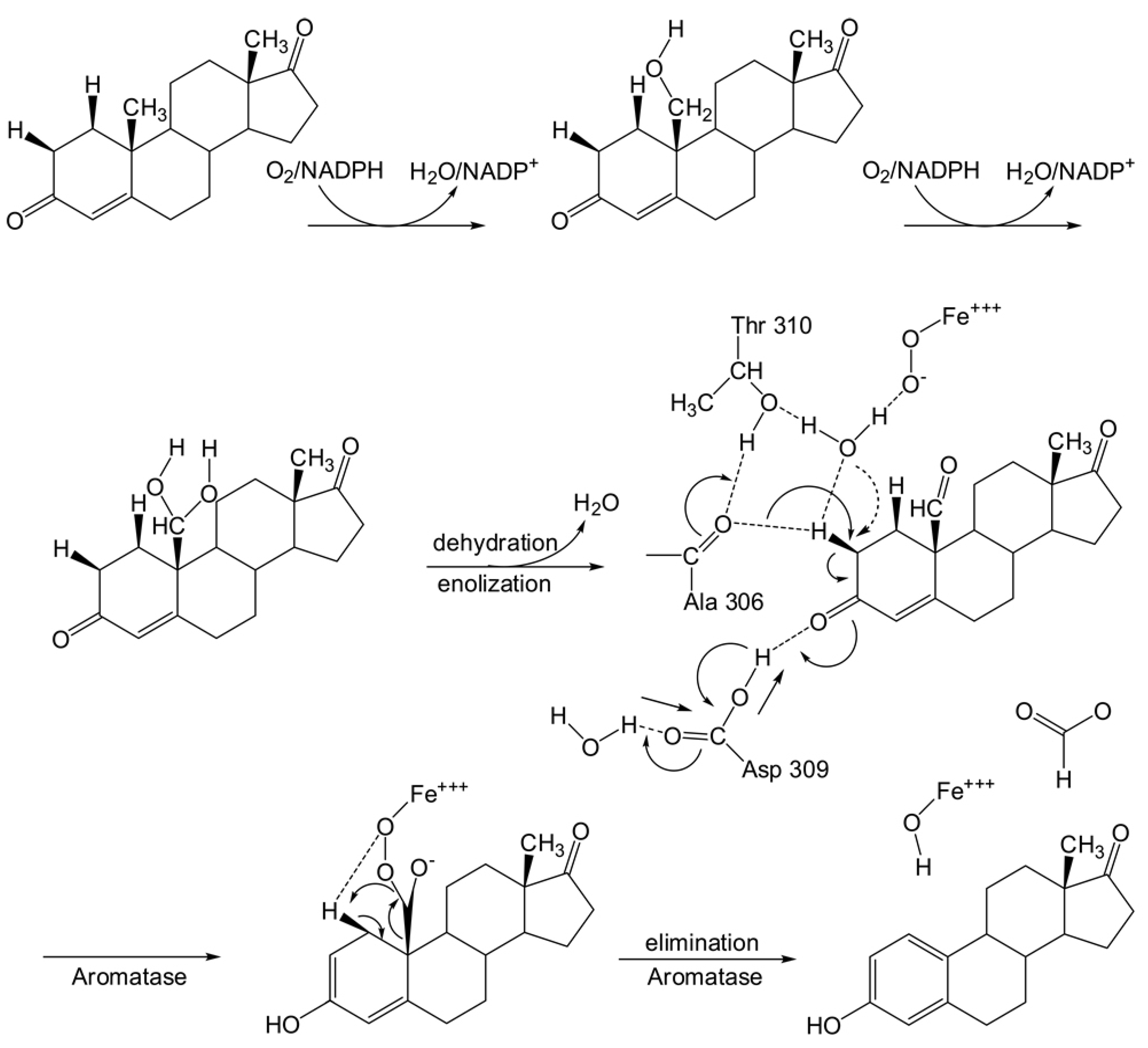
2.7. Mechanism of aromatase catalysis
2.8. Comparing Between Substrate and Product of Aromatase Enzyme
3. Experimental
3.1. Preparation of Protein and Ligand Structures
3.2. Molecular dynamics simulation
3.3. Molecular Docking
3.4. Post-docking analysis
4. Conclusions
Acknowledgements
References
- Fontham, E.T.H.; Thun, M.J.; Ward, E.; Balch, A.J.; Delancey, J.O.L.; Samet, J.M. American Cancer Society perspectives on environmental factors and cancer. CA Cancer J. Clin. 2009, 59, 343–351. [Google Scholar] [CrossRef]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45 (Suppl.), S116–S124. [Google Scholar] [CrossRef]
- Russo, J.; Lareef, M.H.; Balogh, G.; Guo, S.; Russo, I.H. Estrogen and its metabolites are carcinogenic agents in human breast epithelial cells. J. Steroid. Biochem. Mol. Biol. 2003, 87, 1–25. [Google Scholar] [CrossRef]
- Eisen, A.; Trudeau, M.; Shelley, W.; Messersmith, H.; Pritchard, K.I. Aromatase inhibitors in adjuvant therapy for hormone receptor positive breast cancer: A systematic review. Cancer Treat. Rev. 2008, 34, 157–174. [Google Scholar] [CrossRef]
- Santen, R.J.; Santner, S.; Davis, B. Aminoglutethimide inhibits extraglandular estrogen production in postmenopausal women with breast carcinoma. J. Clin. Endocrinol. Metab. 1978, 47, 1257–1265. [Google Scholar] [CrossRef]
- Graves, P.E.; Salhanick, H.A. Stereoselective inhibition of aromatase by enantiomers of aminoglutethimide. Endocrinology 1979, 105, 52–57. [Google Scholar] [CrossRef]
- Santen, R.J.; Misbin, R.I. Aminoglutethimide: Review of pharmacology and clinical use. Pharmacotherapy 1981, 1, 95–120. [Google Scholar]
- Santen, R.J.; Worgul, T.J.; Lipton, A. Aminoglutethimide as treatment of postmenopausal women with advanced breast carcinoma. Ann. Intern. Med. 1982, 96, 94–101. [Google Scholar]
- Hughes, S.W.; Burley, D.M. Aminoglutethimide: a "side-effect" turned to therapeutic advantage. Postgrad. Med. J. 1970, 46, 409–416. [Google Scholar] [CrossRef]
- Demers, L.M.; Boucher, A.E.; Santen, R.J. Aminoglutethimide therapy in breast cancer: Relationship of blood levels to drug-related side effects. Clin. Physiol. Biochem. 1987, 5, 287–291. [Google Scholar]
- Santen, R.J.; Samojlik, E.; Wells, S.A. Resistance of the ovary to blockade of aromatization with aminoglutethimide. J. Clin. Endocrinol. Metab. 1980, 51, 473–477. [Google Scholar] [CrossRef]
- Beretta, K.R.; Hoeffken, K.; Kvinnsland, S.; Trunet, P.; Chaudri, H.A.; Bhatnagar, A.S.; Goldhirsch, A.; Cavalli, F. CGS 16949A, a new aromatase inhibitor in the treatment of breast cancer - A phase I study. Ann. Oncol. 1990, 1, 421–426. [Google Scholar]
- Brueggemeier, R.W.; Hackett, J.C.; Diaz-Cruz, E.S. Aromatase inhibitors in the treatment of breast cancer. Endocrinol. Rev. 2005, 26, 331–345. [Google Scholar] [CrossRef]
- Cunningham, D.; Powles, T.J.; Dowsett, M.; Hutchison, G.; Brodie, A.M.H.; Ford, H.T.; Gazet, J.C.; Coombes, R.C. Oral 4-hydroxyandrostenedione, a new endocrine treatment for disseminated breast cancer. Cancer Chemother. Pharm. 1987, 20, 253–255. [Google Scholar] [CrossRef]
- Plourde, P.V.; Dyroff, M.; Dowsett, M.; Demers, L.; Yates, R.; Webster, A. ARIMIDEXTM: A new oral, once-a-day aromatase inhibitor. J. Steroid Biochem. Mol. Biol. 1995, 53, 175–179. [Google Scholar] [CrossRef]
- Dutta, U.; Pant, K. Aromatase inhibitors: Past, present and future in breast cancer therapy. Med. Oncol. 2008, 25, 113–124. [Google Scholar] [CrossRef]
- Gueto, C.; Torres, J.; Vivas-Reyes, R. CoMFA, LeapFrog and blind docking studies on sulfonanilide derivatives acting as selective aromatase expression regulators. Eur. J. Med. Chem. 2009, 44, 3445–3451. [Google Scholar]
- Thurlimann, B.; Hess, D.; Koberle, D.; Senn, I.; Ballabeni, P.; Pagani, O.; Perey, L.; Aebi, S.; Rochlitz, C.; Goldhirsch, A. Anastrozole ('Arimidex') versus tamoxifen as first-line therapy in postmenopausal women with advanced breast cancer: results of the double-blind cross-over SAKK trial 21/95--a sub-study of the TARGET (Tamoxifen or 'Arimidex' Randomized Group Efficacy and Tolerability) trial. Breast Cancer Res. Treat. 2004, 85, 247–254. [Google Scholar]
- Akhtar, M.; Njar, V.C.O.; Wright, J.N. Mechanistic studies on aromatase and related C-C bond cleaving P-450 enzymes. J. Steroid Biochem. 1993, 44, 375–387. [Google Scholar] [CrossRef]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–223. [Google Scholar]
- Wang, X.; Chen, S. Aromatase destabilizer: novel action of exemestane, a food and drug administration-approved aromatase inhibitor. Cancer Res. 2006, 66, 10281–10286. [Google Scholar] [CrossRef]
- Karkola, S.; Wahala, K. The binding of lignans, flavonoids and coumestrol to CYP450 aromatase: a molecular modelling study. Mol. Cell Endocrinol. 2009, 301, 235–244. [Google Scholar] [CrossRef]
- Loge, C.; Le Borgne, M.; Marchand, P.; Robert, J.M.; Le Baut, G.; Palzer, M.; Hartmann, R.W. Three-dimensional model of cytochrome P450 human aromatase. J. Enzyme. Inhib. Med. Chem. 2005, 581–585. [Google Scholar]
- Paoletta, S.; Steventon, G.B.; Wildeboer, D.; Ehrman, T.M.; Hylands, P.J.; Barlow, D.J. Screening of herbal constituents for aromatase inhibitory activity. Bioorg. Med. Chem. 2008, 16, 8466–8470. [Google Scholar] [CrossRef] [Green Version]
- Nagar, S.; Islam, M.A.; Das, S.; Mukherjee, A.; Saha, A. Pharmacophore mapping of flavone derivatives for aromatase inhibition. Mol. Divers. 2008, 12, 65–76. [Google Scholar] [CrossRef]
- Ariazi, E.A.; Leitão, A.; Oprea, T.I.; Chen, B.; Louis, T.; Bertucci, A.M.; Sharma, C.G. N.; Gill, S.D.; Kim, H.R.; Shupp, H.A.; Pyle, J.R.; Madrack, A.; Donato, A.L.; Cheng, D.; Paige, J.R.; Jordan, V.C. Exemestane's 17-hydroxylated metabolite exerts biological effects as an androgen. Mol. Cancer Ther. 2007, 6, 2817–2827. [Google Scholar]
- Nantasenamat, C.; Naenna, T.; Isarankura Na-Ayudhya, C.; Prachayasittikul, V. Quantitative prediction of imprinting factor of molecularly imprinted polymers by artificial neural network. J. Comput. Aid. Mol. Des. 2005, 19, 509–524. [Google Scholar] [CrossRef]
- Piacham, T.; Isarankura-Na-Ayudhya, C.; Nantasenamat, C.; Yainoy, S.; Ye, L.; Bülow, L.; Prachayasittikul, V. MetalloantibioticMn(II)-bacitracin complex mimicking manganese superoxide dismutase. Biochem. Biophys. Res. Commun. 2006, 341, 925–930. [Google Scholar] [CrossRef]
- Prachayasittikul, V.; Isarankura-Na-Ayudhya, C.; Tantimongcolwat, T.; Nantasenamat, C.; Galla, H.J. EDTA-induced membrane fluidization and destabilization: Biophysical studies on artificial lipid membranes. Acta. Biochim. Biophys. Sin. 2007, 39, 901–913. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. Quantitative structure-imprinting factor relationship of molecularly imprinted polymers. Biosens. Bioelectron. 2007, 22, 3309–3317. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Tansila, N.; Naenna, T.; Prachayasittikul, V. Prediction of GFP spectral properties using artificial neural network. J. Comput. Chem. 2007, 28, 1275–1289. [Google Scholar] [CrossRef]
- Suksrichavalit, T.; Prachayasittikul, S.; Piacham, T.; Isarankura-Na-Ayudhya, C.; Nantasenamat, C.; Prachayasittikul, V. Copper complexes of nicotinic-aromatic carboxylic acids as superoxide dismutase mimetics. Molecules 2008, 13, 3040–3056. [Google Scholar] [CrossRef]
- Isarankura-Na-Ayudhya, C.; Nantasenamat, C.; Buraparuangsang, P.; Piacham, T.; Ye, L.; Bülow, L.; Prachayasittikul, V. Computational insights on sulfonamide imprinted polymers. Molecules 2008, 13, 3077–3091. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. Prediction of bond dissociation enthalpy of antioxidant phenols by support vector machine. J. Mol. Graph. Model. 2008, 27, 188–196. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Piacham, T.; Tantimongcolwat, T.; Naenna, T.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. QSAR model of the quorum-quenching N-acyl-homoserinelactonelactonase activity. J. Biol. Syst. 2008, 16, 279–293. [Google Scholar] [CrossRef]
- Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. A practical overview of quantitative structure-activity relationship. EXCLI J. 2009, 74–88. [Google Scholar]
- Suksrichavalit, T.; Prachayasittikul, S.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Copper complexes of pyridine derivatives with superoxide scavenging and antimicrobial activities. Eur. J. Med. Chem. 2009, 44, 3259–3265. [Google Scholar] [CrossRef]
- Piacham, T.; Nantasenamat, C.; Suksrichavalit, T.; Puttipanyalears, C.; Pissawong, T.; Maneewas, S.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Synthesis and theoretical study of molecularly imprinted nanospheres for recognition of tocopherols. Molecules 2009, 14, 2985–3002. [Google Scholar] [CrossRef]
- Thippakorn, C.; Suksrichavalit, T.; Nantasenamat, C.; Tantimongcolwat, T.; Isarankura-Na-Ayudhya, C.; Naenna, T.; Prachayasittikul, V. Modeling the LPS neutralization activity of anti-endotoxins. Molecules 2009, 14, 1869–1888. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Nantasenamat, C.; Naenna, T.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Modeling the activity of furin inhibitors using artificial neural network. Eur. J. Chem. 2009, 44, 1664–1673. [Google Scholar]
- Prachayasittikul, S.; Wongsawatkul, O.; Worachartcheewan, A.; Nantasenamat, C.; Ruchirawat, S.; Prachayasittikul, V. Elucidating the Structure-Activity relationships of the vasorelaxation and antioxidation properties of thionicotinic acid derivatives. Molecules 2010, 15, 198–214. [Google Scholar] [CrossRef]
- Diller, D.J.; Li, R. Kinases, Homology Models, and High Throughput Docking. J. Med. Chem. 2003, 46, 4638–4647. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. Docking and 3D-QSAR studies of diverse classes of human aromatase (CYP19) inhibitors. J. Mol. Model. 2010, 1597–1616. [Google Scholar]
- Laskowski, R.A.; Hutchinson, E.G.; Michie, A.D.; Wallace, A.C.; Jones, M.L.; Thornton, J.M. PDBsum: A Web-based database of summaries and analyses of all PDB structures. Trends. Biochem. Sci. 1997, 22, 488–490. [Google Scholar] [CrossRef]
- Ni, C.Z.; Li, C.; Wu, J.C.; Spada, A.P.; Ely, K.R. Conformational restrictions in the active site of unliganded human caspase-3. J. Mol. Recognit. 2003, 16, 121–124. [Google Scholar] [CrossRef]
- Becker, J.W.; Rotonda, J.; Soisson, S.M.; Aspiotis, R.; Bayly, C.; Francoeur, S.; Gallant, M.; Garcia-Calvo, M.; Giroux, A.; Grimm, E.; Han, Y.; McKay, D.; Nicholson, D.W.; Peterson, E.; Renaud, J.; Roy, S.; Thornberry, N.; Zamboni, R. Reducing the Peptidyl Features of Caspase-3 Inhibitors: A Structural Analysis. J. Med. Chem. 2004, 47, 2466–2474. [Google Scholar]
- De Graaf, C.; Oostenbrink, C.; Keizers, P.H.J.; Van Der Wijst, T.; Jongejan, A.; Vermeulen, N.P.E. Catalytic site prediction and virtual screening of cytochrome P450 2D6 substrates by consideration of water and rescoring in automated docking. J. Med. Chem. 2006, 49, 2417–2430. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Hong, Y.; Cho, M.; Yuan, Y.C.; Chen, S. Molecular basis for the interaction of four different classes of substrates and inhibitors with human aromatase. Biochem. Pharmacol. 2008, 75, 1161–1169. [Google Scholar]
- Hong, Y.; Li, H.; Yuan, Y.-C.; Chen, S. Molecular Characterization of Aromatase. Ann. NY. Acad. Sci. 2009, 1155, 112–120. [Google Scholar]
- Goss, P.E.; Strasser, K. Aromatase inhibitors in the treatment and prevention of breast cancer. J. Clin. Oncol. 2001, 19, 881–894. [Google Scholar]
- Geisler, J.; Lønning, P.E. Endocrine effects of aromatase inhibitors and inactivators in vivo: Review of data and method limitations. J. Steroid Biochem Mol. Biol. 2005, 95, 75–81. [Google Scholar] [CrossRef]
- Feuillan, P.P.; Jones, J.; Cutler, G.B., Jr. Long term testolactone therapy for precocious puberty in girls with the McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 647–651. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Houk, K.N. Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene. J. Am. Chem. Soc. 2008, 130, 10854–10855. [Google Scholar] [CrossRef]
- Favia, A.D.; Cavalli, A.; Masetti, M.; Carotti, A.; Recanatini, M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinato cytochromes. Proteins: Structure Function Genetics 2006, 62, 1074–1087. [Google Scholar] [CrossRef]
- Neves, M.A.C.; Dinis, T.C.P.; Colombo, G.; Sá e Melo, M.L. Fast three dimensional pharmacophore virtual screening of new potent Non-steroid aromatase inhibitors. J. Med. Chem. 2009, 52, 143–150. [Google Scholar] [CrossRef]
- Buzdar, A.U.; Robertson, J.F.; Eiermann, W.; Nabholtz, J.M. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer 2002, 95, 2006–2016. [Google Scholar] [CrossRef]
- Wolf, L.K. PyRx. C&EN. 2009, 87, 31. [Google Scholar]
- Favia, A.D.; Cavalli, A.; Masetti, M.; Carotti, A.; Recanatini, M. Three-dimensional model of the human aromatase enzyme and density functional parameterization of the iron-containing protoporphyrin IX for a molecular dynamics study of heme-cysteinatocytochromes. Proteins 2006, 62, 1074–1087. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J. A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2003. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Krieger, E.; Nabuurs, S.B.; Vriend, G. Homology modeling. Meth. Biochem. Anal. 2003, 44, 509–523. [Google Scholar]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA - A self-parameterizing force field. Protein. Struct. Funct. Genet. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M., Jr.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle meshEwald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Solis, F.J.; Wets, R.J.B. Minimization by random search techniques. Math.Oper. Res. 1981, 6, 19–30. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Prot. Bioinf. 2008, 24. 8.14.1-8.14.40. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 15–26. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597-3617. https://doi.org/10.3390/molecules16053597
Suvannang N, Nantasenamat C, Isarankura-Na-Ayudhya C, Prachayasittikul V. Molecular Docking of Aromatase Inhibitors. Molecules. 2011; 16(5):3597-3617. https://doi.org/10.3390/molecules16053597
Chicago/Turabian StyleSuvannang, Naravut, Chanin Nantasenamat, Chartchalerm Isarankura-Na-Ayudhya, and Virapong Prachayasittikul. 2011. "Molecular Docking of Aromatase Inhibitors" Molecules 16, no. 5: 3597-3617. https://doi.org/10.3390/molecules16053597
APA StyleSuvannang, N., Nantasenamat, C., Isarankura-Na-Ayudhya, C., & Prachayasittikul, V. (2011). Molecular Docking of Aromatase Inhibitors. Molecules, 16(5), 3597-3617. https://doi.org/10.3390/molecules16053597