The Effects of Co-Treatment of 9-cis-Retinoic Acid and 15-Deoxy-Δ (12,14)-prostaglandin J2 on Microglial Activation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Aβ/IFNγ-induced microglial immune response was suppressed by the co-treatment of RA and 15d-PGJ2
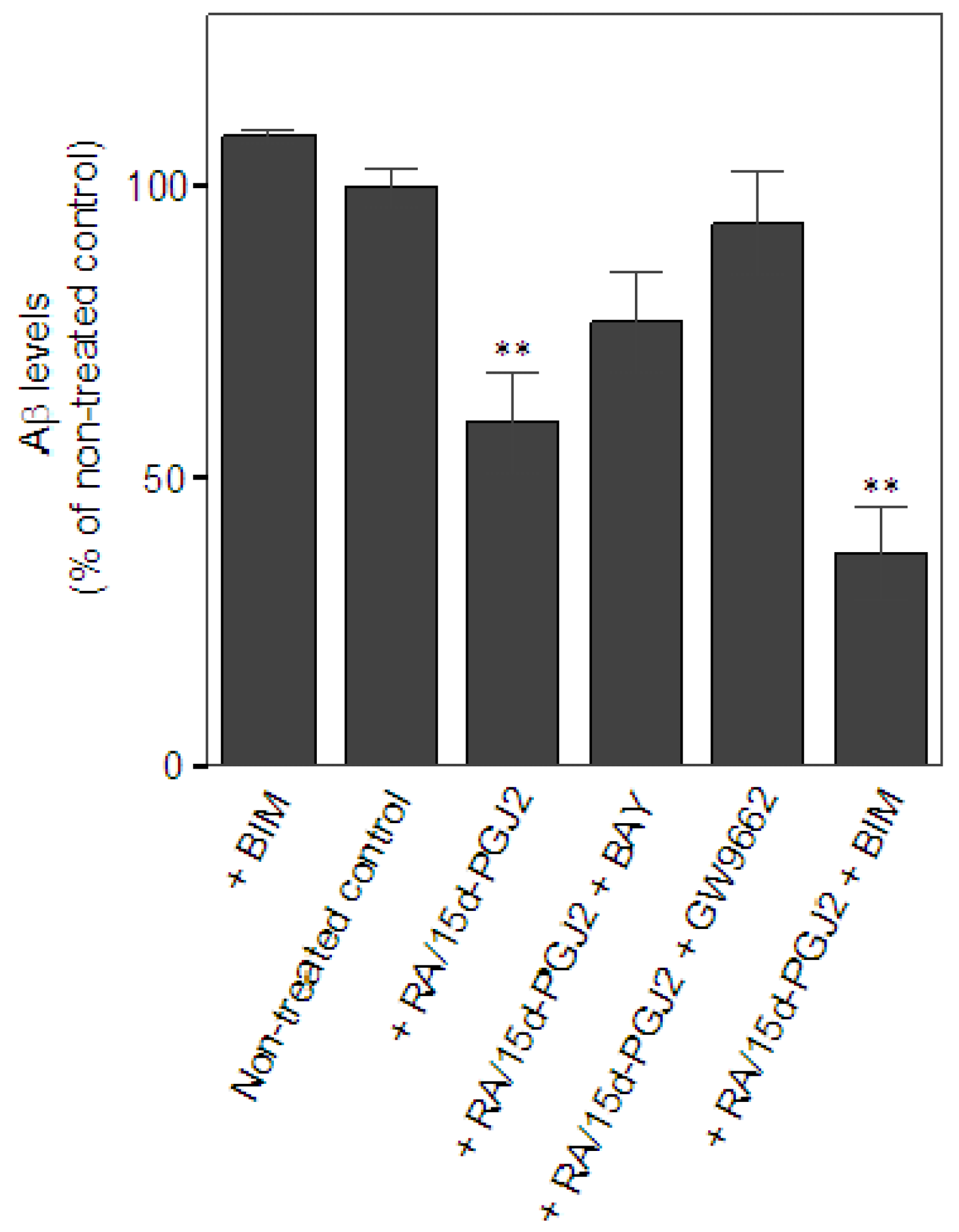
2.2. Co-treatment of RA and 15d-PGJ2 enhanced microglial Aβ clearance
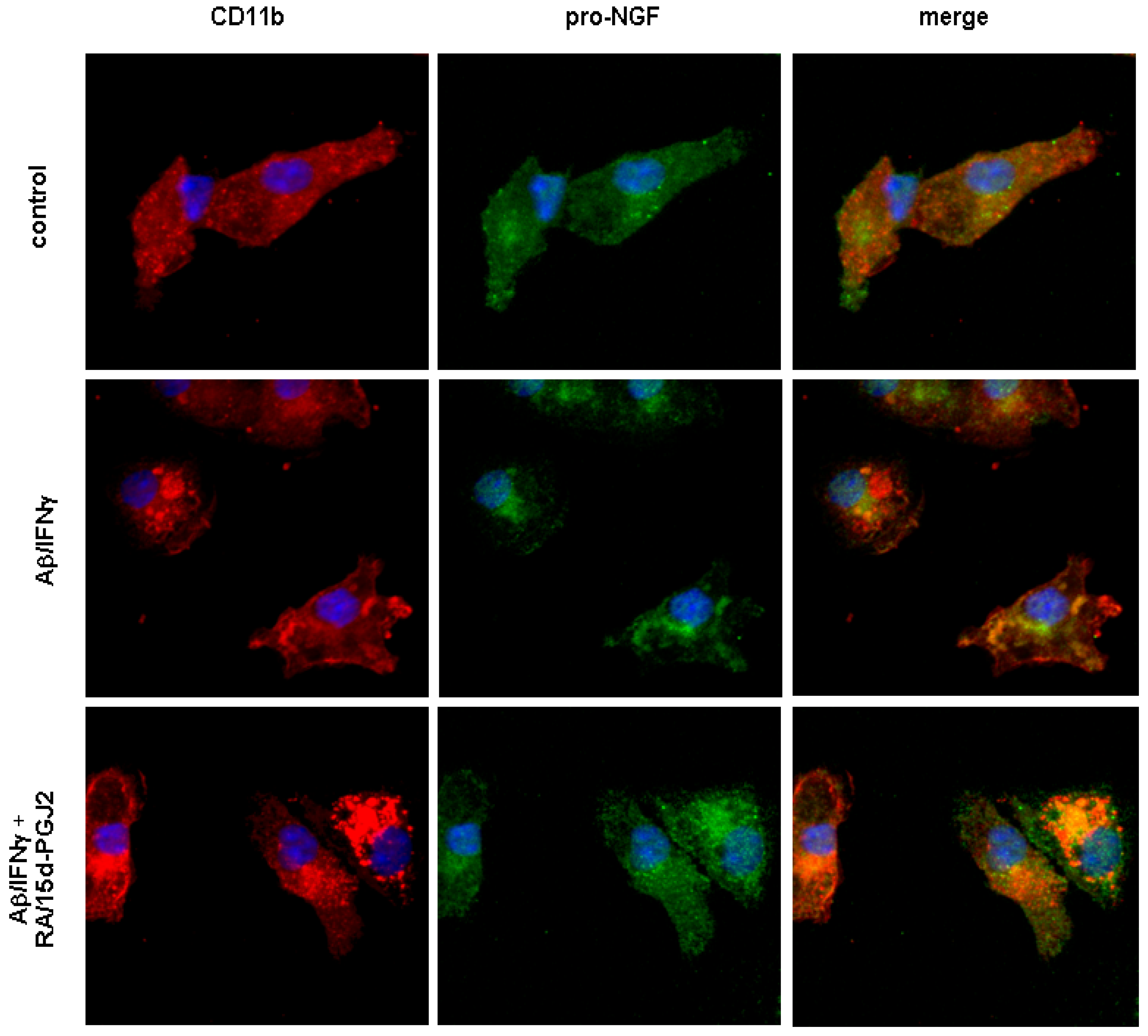
2.3. Aβ treatment inhibited the secretion of microglial pro-NGF independent of the status of microglial activation
3. Experimental
3.1. Animals and materials
3.2. Primary microglial cultures
3.3. Aβ-induced immune activation
3.4. Aβ clearance assay
3.5. Pro-NGF measurement
3.6. Cell viability assay
3.7. Statistics
4. Conclusions
Acknowledgements
References
- Cooper, N.R.; Kalaria, R.N.; McGeer, P.L.; Rogers, J. Key issues in Alzheimer’s disease inflammation. Neurobiol. Aging 2000, 21, 451–453. [Google Scholar] [CrossRef]
- Lue, L.F.; Rydel, R.; Brigham, E.F.; Yang, L.B.; Hampel, H.; Murphy, G.M., Jr.; Brachova, L.; Yan, S.D.; Walker, D.G.; Shen, Y.; Rogers, J. Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 2001, 35, 72–79. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. Inflammation, autotoxicity and Alzheimer disease. Neurobiol. Aging 2001, 22, 799–809. [Google Scholar] [CrossRef]
- Streit, W.J. Microglia and Alzheimer’s disease pathogenesis. J Neurosci. Res. 2004, 77, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, N.J.; Decorti, F.; Pappas, T.C.; Taglialatela, G. Cytokine/neurotrophin interaction in the aged central nervous system. J. Anat. 2000, 197, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Montine, T.J.; Morrow, J.D. Fatty acid oxidation in the pathogenesis of Alzheimer’s disease. Am. J. Pathol. 2005, 166, 1283–1289. [Google Scholar] [CrossRef]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L. Diffusible, nonfibrillar ligands derived from A 1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural. Transm. 2010, 117, 919–947. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.J.; Cai, Y.; Lungo, W.; Fu, P.; Locker, J.; French, S.; Sucov, H.M. Peroxisome proliferator-activated receptor alpha-mediated pathways are altered in hepatocyte-specific retinoid X receptor alpha-deficient mice. J. Biol. Chem. 2000, 275, 28285–28290. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARS in health and disease. Nature 2000, 6785, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A. Peroxisome proliferator-activated receptors and the regulation of mammalian lipid metabolism. Biochem. Soc. Trans. 2002, 30, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Wagner, J.A. Physiological and therapeutic roles of peroxisome proliferator-activated receptors. Diabetes Technol. Ther. 2002, 4, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Blanquart, C.; Barbier, O.; Fruchart, J.C.; Staels, B.; Glineur, C. Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 2003, 85, 267–273. [Google Scholar] [CrossRef]
- Combs, C.K.; Johnson, D.E.; Karlo, J.C.; Cannady, S.B.; Landreth, G.E. Inflammatory mechanisms in Alzheimer’s disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J. Neurosci. 2000, 20, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; Kang, D.E.; Marquez-Sterling, N.; Golde, T.E.; Koo, E.H. A subset of NSAIDs lower amyloidogenic Aβ 42 independently of cyclooxygenase activity. Nature. 2001, 414, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Ongini, E.; Wenk, G. Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J. Neurochem. 2004, 91, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Hanke, A.; Dewachter, I.; Kuiperi, C.; O’Banion, K.; Klockgether, T.; Van Leuven, F.; Landreth, G.E. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 2005, 128, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Cabrero, A.; Laguna, J.C.; Vazquez, M. Peroxisome proliferator-activated receptors and the control of inflammation. Curr. Drug Targets Inflamm. Allergy 2002, 1, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kainu, T.; Wikstrom, A.C.; Gustafsson, J.A.; Pelto-Huikko, M. Localization of the peroxisome proliferator-activated receptor in the brain. Neuroreport 1994, 5, 2481–2485. [Google Scholar] [CrossRef] [PubMed]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis Retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Lefebvre, P. Molecular basis for designing selective modulators of retinoic acid receptor transcriptional activities. Curr. Drug Targets Immune. Endocr. Metabol. Disord. 2001, 1, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Petrova, T.V.; Akama, K.T.; Van Eldik, L.J. Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc. Natl. Acad. Sci. USA 1999, 96, 4668–4673. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kwon, K.J.; Park, J.Y.; Lee, S.H.; Moon, C.H.; Baik, E.J. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: associated with iNOS and COX-2. Brain Res. 2002, 941, 1–10. [Google Scholar] [CrossRef]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.E. Changes in the immune system in depression and dementia: causal or co-incidental effects? Int. J. Dev. Neurosci. 2001, 19, 305–312. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Frade, J.M.; Barde, Y.A. Identify microglial cells as the source of NGF as a killing agent in the developing eye. Neuron 1998, 20, 35–41. [Google Scholar] [CrossRef]
- Barker, P.A. p75NTR is positively promiscuous: novel partners and new insights. Neuron 2004, 42, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Fahnestock, M.; Michalski, B.; Xu, B.; Coughlin, M.D. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer’s disease. Mol. Cell. Neurosci. 2001, 18, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, C.E.; Podlesniy, P.; Vidal, N.; Arevalo, J.C.; Lee, R.; Hempstead, B.; Ferrer, I.; Iglesias, M.; Espinet, C. Pro-NGF isolated from the human brain affected by Alzheimer’s disease induces neuronal apoptosis mediated by p75NTR. Am. J. Pathol. 2005, 166, 533–543. [Google Scholar] [CrossRef]
- Coulson, E.J. Does the p75 neurotrophin receptor mediate Abeta-induced toxicity in Alzheimer’s disease? J Neurochem. 2006, 98, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Shie, F.S.; Breyer, R.M.; Montine, T.J. Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am. J. Pathol. 2005, 166, 1163–1172. [Google Scholar] [CrossRef]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar beta-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Schor, N.F. The p75 neurotrophin receptor in human development and disease. Prog. Neurobiol. 2005, 77, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Kichev, A.; Ilieva, E.V; Piñol-Ripoll, G.; Podlesniy, P.; Ferrer, I.; Portero-Otín, M.; Pamplona, R.; Espinet, C. Cell death and learning impairment in mice caused by in vitro modified pro-NGF can be related to its increased oxidative modifications in Alzheimer disease. Am. J. Pathol. 2009, 175, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D. Modulation of microglial activation state following passive immunization in amyloid depositing transgenic mice. Neurochem. International. 2006, 49, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, D.S.; DaSilva, K.; Fenili, D.; St George-Hyslop, P.; McLaurin, J. Immunotherapy for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 14657–14662. [Google Scholar] [CrossRef] [PubMed]
- García-Bueno, B.; Caso, J.R.; Pérez-Nievas, B.G.; Lorenzo, P.; Leza, J.C. Effects of Peroxisome Proliferator-Activated Receptor Gamma Agonists on Brain Glucose and Glutamate Transporters after Stress in Rats. Neuropsychopharmacology 2007, 32, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Dicou, E. Multiple biological activities for two peptides derived from the nerve growth factor precursor. Biochem. Biophys. Res. Commun. 2006, 347, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Jaffar, S.; Counts, S.E.; Ma, S.Y.; Dadko, E.; Gordon, M.N.; Morgan, D.; Mufson, E.J. Neuropathology of mice carrying mutant APP(swe) and/or PS1(M146L) transgenes: alterations in the p75(NTR) cholinergic basal forebrain septohippocampal pathway. Exp. Neurol. 2001, 170, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.A.; Paden, C.M. Upregulation of the p75 low-affinity neurotrophin receptor by phagocytically active perivascular active cells in the rat neural lobe. Cell Tissue Res. 2001, 303, 81–91. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds RA and 15d-PGJ2 are available from Sigma. |




{kind=link}
{kind=link}
{kind=link}
| iNOS | COX-2 | |
|---|---|---|
| Aβ/IFNγ + RA/15d-PGJ2 | 55.4 ± 30.3%*, n = 5 | 47.2 ± 19.7%**, n = 5 |
| Aβ/IFNγ + RA/15d-PGJ2 + GW9662 | 51.4 ± 27.3%*, n = 5 | 60.3 ± 24.7%**, n = 5 |
| Aβ/IFNγ + RA/15d-PGJ2 + BAY | 56.6 ± 29.1%*, n = 6 | 48.8 ± 19.3%**, n = 6 |
| Aβ/IFNγ + RA/15d-PGJ2 + BIM | 44.0 ± 9.5%**, n = 4 | 46.4 ± 12.0%**, n = 4 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hsu, P.-C.; Tsay, H.-J.; Montine, T.J.; Shie, F.-S. The Effects of Co-Treatment of 9-cis-Retinoic Acid and 15-Deoxy-Δ (12,14)-prostaglandin J2 on Microglial Activation. Molecules 2011, 16, 4045-4058. https://doi.org/10.3390/molecules16054045
Hsu P-C, Tsay H-J, Montine TJ, Shie F-S. The Effects of Co-Treatment of 9-cis-Retinoic Acid and 15-Deoxy-Δ (12,14)-prostaglandin J2 on Microglial Activation. Molecules. 2011; 16(5):4045-4058. https://doi.org/10.3390/molecules16054045
Chicago/Turabian StyleHsu, Pei-Chien, Huey-Jen Tsay, Thomas J. Montine, and Feng-Shiun Shie. 2011. "The Effects of Co-Treatment of 9-cis-Retinoic Acid and 15-Deoxy-Δ (12,14)-prostaglandin J2 on Microglial Activation" Molecules 16, no. 5: 4045-4058. https://doi.org/10.3390/molecules16054045
APA StyleHsu, P. -C., Tsay, H. -J., Montine, T. J., & Shie, F. -S. (2011). The Effects of Co-Treatment of 9-cis-Retinoic Acid and 15-Deoxy-Δ (12,14)-prostaglandin J2 on Microglial Activation. Molecules, 16(5), 4045-4058. https://doi.org/10.3390/molecules16054045