Synthetic Studies on Bioactive Natural Polyketides: Intramolecular Nitrile Oxide-Olefin Cycloaddition Approach for Construction of a Macrolactone Skeleton of Macrosphelide B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
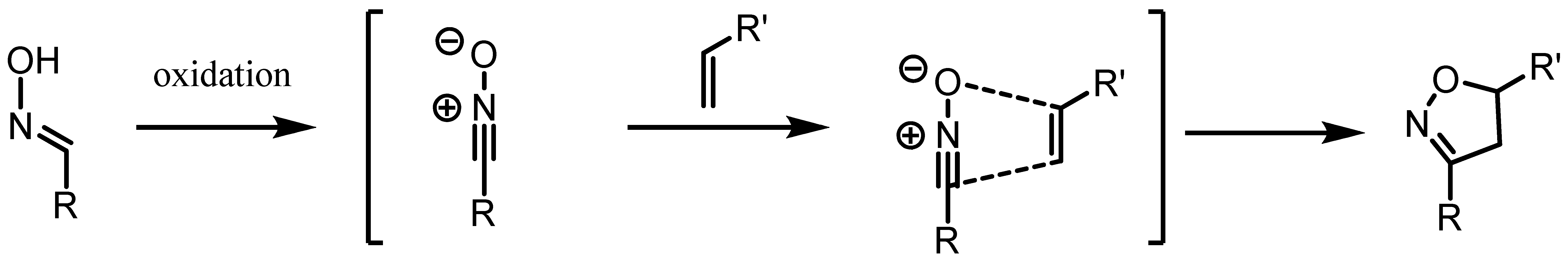
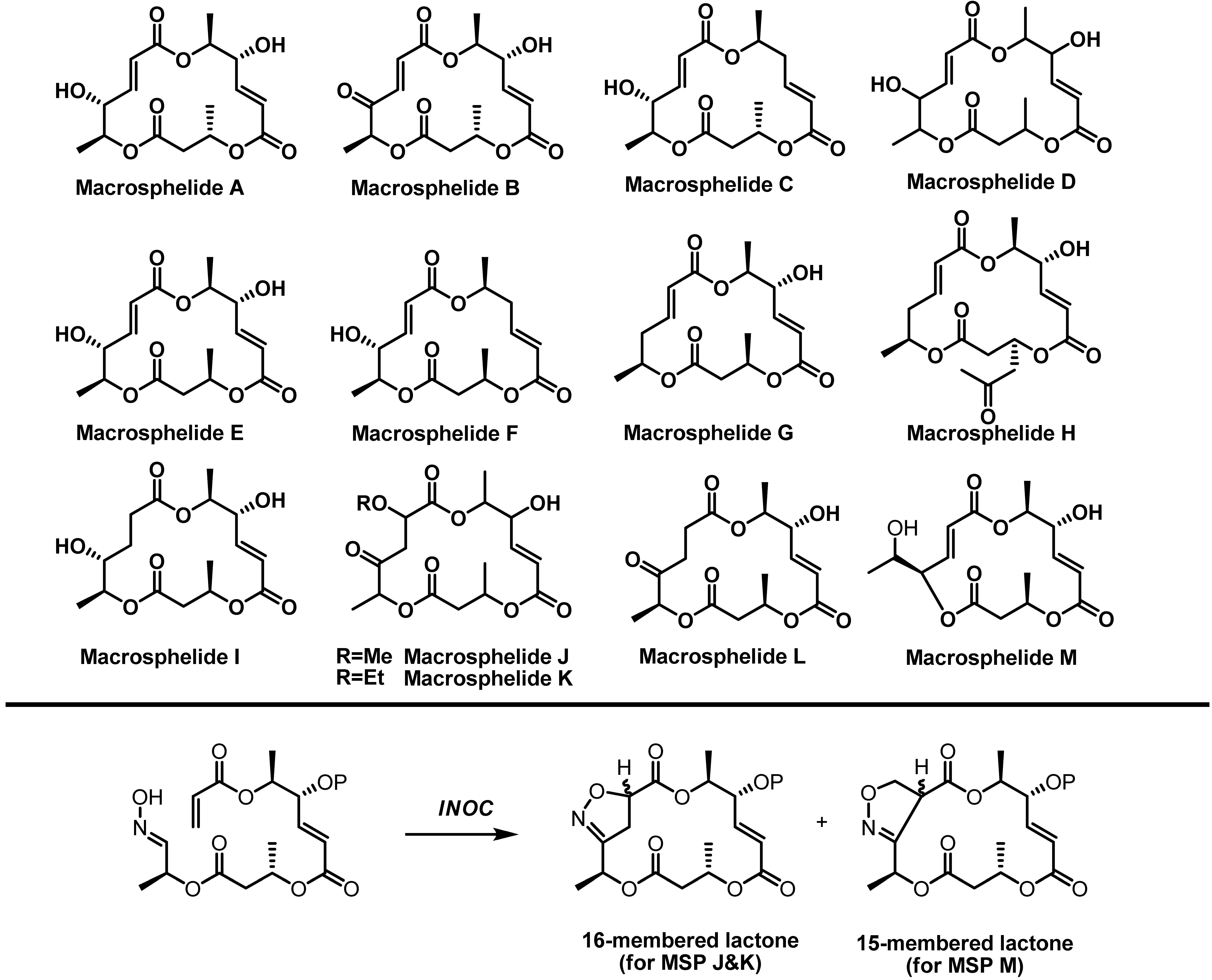
:1. Introduction

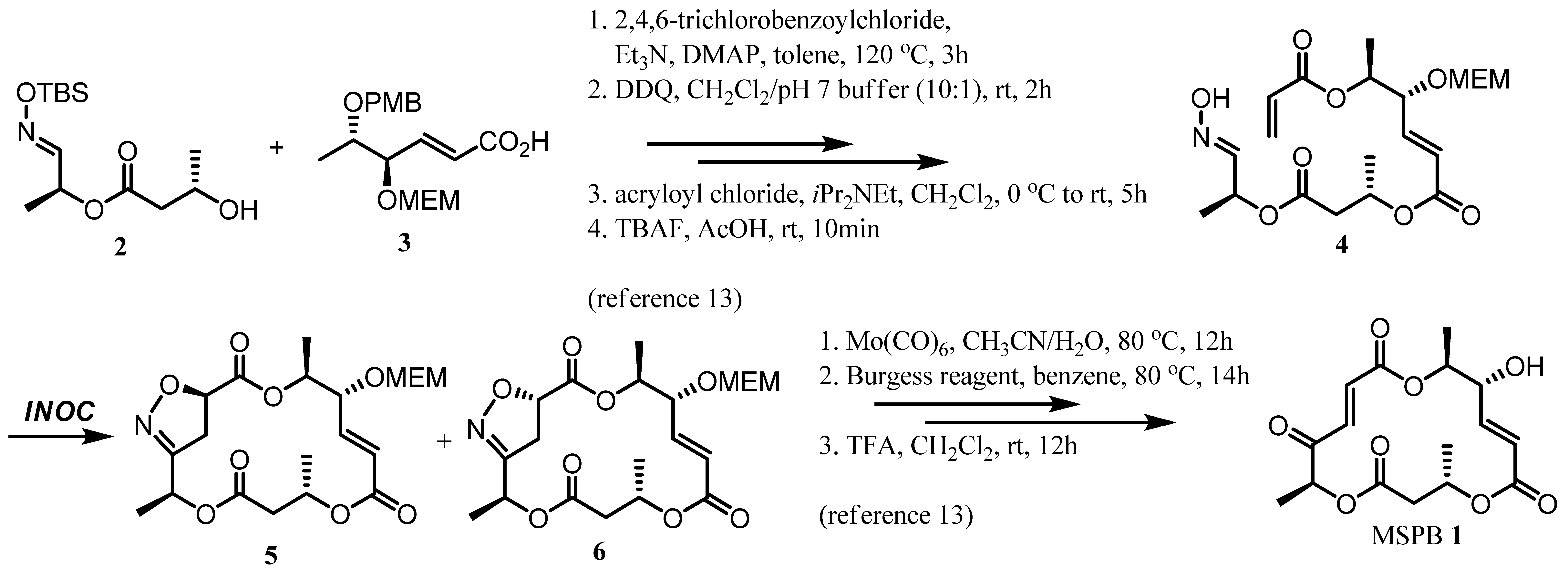
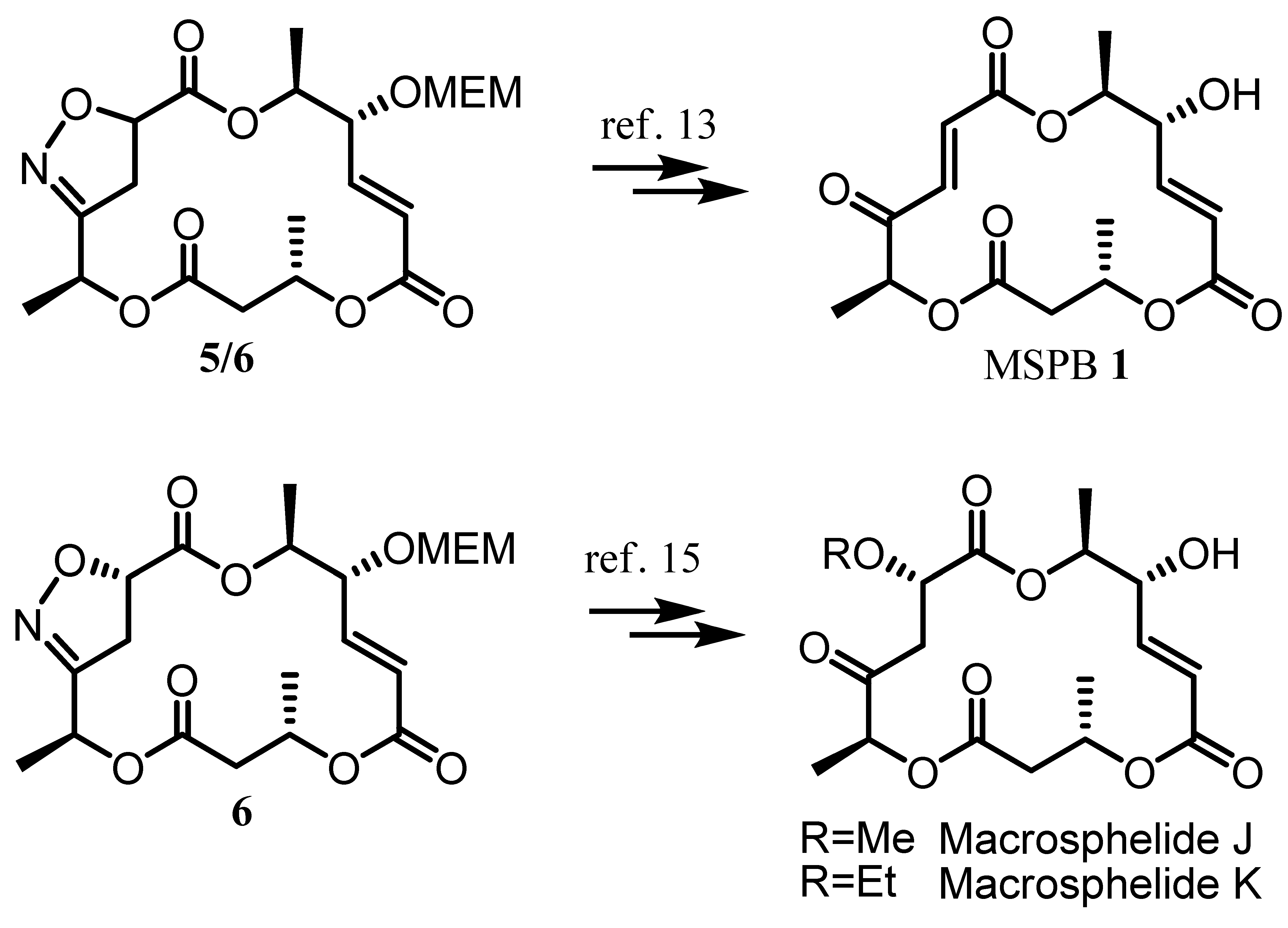
2. Results and Discussion





3. Experimental
3.1. General
3.2. Representative INOC Procedure
4. Conclusions
Acknowledgements
References and Notes
- Dewick, P.M. Medicinal Natural Products, 2nd ed; Wiley and Sons: Sussex, UK, 2001. [Google Scholar]
- Kang, E.J.; Lee, E. Total synthesis of oxacyclic macrodiolide natural products. Chem. Rev. 2005, 105, 4343–4378. [Google Scholar]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 505–509. [Google Scholar]
- Nicolaou, K.C.; Chakraborty, T.K.; Piscopio, A.D.; Minowa, N.; Bertinato, P. Total synthesis of rapamycin. J. Am. Chem. Soc. 1993, 115, 4419–4420. [Google Scholar] [CrossRef]
- Grubbs, R.H.; Miller, S.J.; Fu, G.C. Ring-closing metathesis and related processes in organic synthesis. Acc. Chem. Res. 1995, 28, 446–452. [Google Scholar] [CrossRef]
- Deiters, A.; Martin, S.F. Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis. Chem. Rev. 2004, 104, 2299–2238. [Google Scholar]
- Asaoka, M.; Abe, M.; Mukata, T.; Takei, H. Synthesis of macrocyclic lactones applying intramolecular 1,3-dipolar cycloaddition: synthesis of (+/-)-A26771B. Chem. Lett. 1982, 11, 215–218. [Google Scholar]
- Kim, D.; Lee, J.; Shim, P.; Lim, J.I.; Doi, T.; Kim, S. Role of conformational effects on the regioselectivity of macrocyclic INOC reactions: Two new asymmetric total syntheses of (+)-brefeldin A. J. Org. Chem. 2002, 67, 772–781. [Google Scholar] [CrossRef]
- Gothelf, K.V.; Jorgensen, K.A. Asymmetric 1,3-dipolar cycloaddition. Chem. Rev 1998, 98, 863–910. [Google Scholar] [CrossRef]
- Curran, D.P.; Fenk, C.J. Thermolysis of bis[2=[(trimethylsilyl)oxy]propyl]furoxan (TOP-furoxan). The first practical method for intermolecular cycloaddition of an in situ generated nitrile oxide with 1,2-di- and trisubstituted olefins. J. Am. Chem. Soc. 1985, 107, 6023–6028. [Google Scholar] [CrossRef]
- Sibi, M.P.; Itoh, K.; Jasperse, C.P. Chiral lewis acid catalysis in nitrile oxide cycloadditions. J. Am. Chem. Soc. 2004, 126, 718–719. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Wong, W.F.; Cheng, H.M.; Kwok, W.S.; So, K.H. Intramolecular nitrile oxide−alkene cycloaddition of sugar derivatives with unmasked hydroxyl group(s). Org. Lett. 2007, 9, 753–756, and references cited therein. [Google Scholar] [CrossRef]
- Paek, S.-M.; Seo, S.-Y.; Kim, S.-H.; Jung, J.-W.; Lee, Y.-S.; Jung, J.-K.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef]
- Paek, S.-M.; Yun, H.; Kim, N.-J.; Jung, J.-W.; Chang, D.-J.; Lee, S.; Yoo, J.; Park, H.-J.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B; studies on the macro-ring closure strategy. J. Org. Chem. 2009, 74, 554–561. [Google Scholar]
- Yun, H.; Paek, S.-M.; Jung, J.-W.; Kim, N.-J.; Kim, S.-H.; Suh, Y.-G. First total syntheses of (-)-macrosphelides J and K and elucidation of their absolute configuration. Chem. Commun. 2009, 18, 2463–2465. [Google Scholar]
- Sunazuka, T.; Hirose, T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Ōmura, S.; Sprengeler, P.A.; Smith, A.B., III. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell-cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar]
- Ono, M.; Nakamura, H.; Konno, F.; Akita, H. Total syntheses of macrosphelides (+)-A, (-)-A and (+)-E. Tetrahedron: Asymmetry 2000, 11, 2753–2764. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumar, G.B.; Kurachi, T.; Acharya, H.P.; Yamazaki, T.; Kitazume, T. Furan ring oxidation strategy for the synthesis of macrosphelide A and B. J. Org. Chem. 2001, 66, 2011–2018. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Mouli, C.C. The total synthesis of macrosphelides A and E from carbohydrate precursors. Tetrahedron Lett. 2002, 43, 9159–9161. [Google Scholar] [CrossRef]
- Kusaka, S.-I.; Dohi, S.; Doi, T.; Takahashi, T. Total synthesis of macrosphelide A by way of palladium-catalyzed carbonylative esterification. Tetrahedron Lett. 2003, 44, 8857–8859. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. New strategy for the total synthesis of macrosphelides A and B based on ring-closing metathesis. Org. Lett. 2003, 5, 2939–2941. [Google Scholar] [CrossRef]
- Takahashi, T.; Kusaka, S.-I.; Doi, T.; Sunazuka, T.; Omura, S. A combinatorial synthesis of a macrosphelide library utilizing a palladium-catalyzed carbonylation on a polymer support. Angew. Chem., Int. Ed. 2003, 42, 5230–5234. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Ishihara, K.; Ahmed, K.; Zhao, Q.-L.; Kondo, T.; Nemoto, H. Synthesis of macrosphelides with a thiazole side chain: New antitumor candidates having apoptosis-inducing property. Org. Lett. 2006, 8, 4609–4612. [Google Scholar] [CrossRef]
- Matsuya, Y.; Nemoto, H. Artificial macrosphelides as a novel apoptosis-inducing compound. Heterocycles 2010, 81, 57–66, and references cited therein. [Google Scholar] [CrossRef]
- The regioisomer of the INOC product was not detected. This result matches with the previous observations reported in references [7] and [8].
- Corey, E.J.; Xu, F.; Noe, M.C. A rational approach to catalytic enantioselective enolate alkylation using a structurally rigidified and defined chiral quaternary ammonium salt under phase transfer conditions. J. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar] [CrossRef]
- Park, H.-G.; Jeong, B.-S.; Yoo, M.-S.; Lee, J.-H.; Park, M.-K.; Lee, Y.-J.; Kim, M.-J.; Jew, S.-S. Highly enantioselective and practical cinchona-derived phase-transfer catalysts for the synthesis of α-amino acids. Angew. Chem., Int. Ed. 2002, 41, 3036–3038. [Google Scholar] [CrossRef]
- A manuscript on detailed mechanism of the corresponding INOC and the synthetic application of isoxazoline 6 to the preparation of macrosphelide J & K is in preparation.
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paek, S.-M.; Suh, Y.-G. Synthetic Studies on Bioactive Natural Polyketides: Intramolecular Nitrile Oxide-Olefin Cycloaddition Approach for Construction of a Macrolactone Skeleton of Macrosphelide B. Molecules 2011, 16, 4850-4860. https://doi.org/10.3390/molecules16064850
Paek S-M, Suh Y-G. Synthetic Studies on Bioactive Natural Polyketides: Intramolecular Nitrile Oxide-Olefin Cycloaddition Approach for Construction of a Macrolactone Skeleton of Macrosphelide B. Molecules. 2011; 16(6):4850-4860. https://doi.org/10.3390/molecules16064850
Chicago/Turabian StylePaek, Seung-Mann, and Young-Ger Suh. 2011. "Synthetic Studies on Bioactive Natural Polyketides: Intramolecular Nitrile Oxide-Olefin Cycloaddition Approach for Construction of a Macrolactone Skeleton of Macrosphelide B" Molecules 16, no. 6: 4850-4860. https://doi.org/10.3390/molecules16064850
APA StylePaek, S. -M., & Suh, Y. -G. (2011). Synthetic Studies on Bioactive Natural Polyketides: Intramolecular Nitrile Oxide-Olefin Cycloaddition Approach for Construction of a Macrolactone Skeleton of Macrosphelide B. Molecules, 16(6), 4850-4860. https://doi.org/10.3390/molecules16064850