Synthesis and Spectroscopic Characterization of Two Tetrasubstituted Cationic Porphyrin Derivatives

 and
and
Abstract
:1. Introduction

2. Results and Discussion
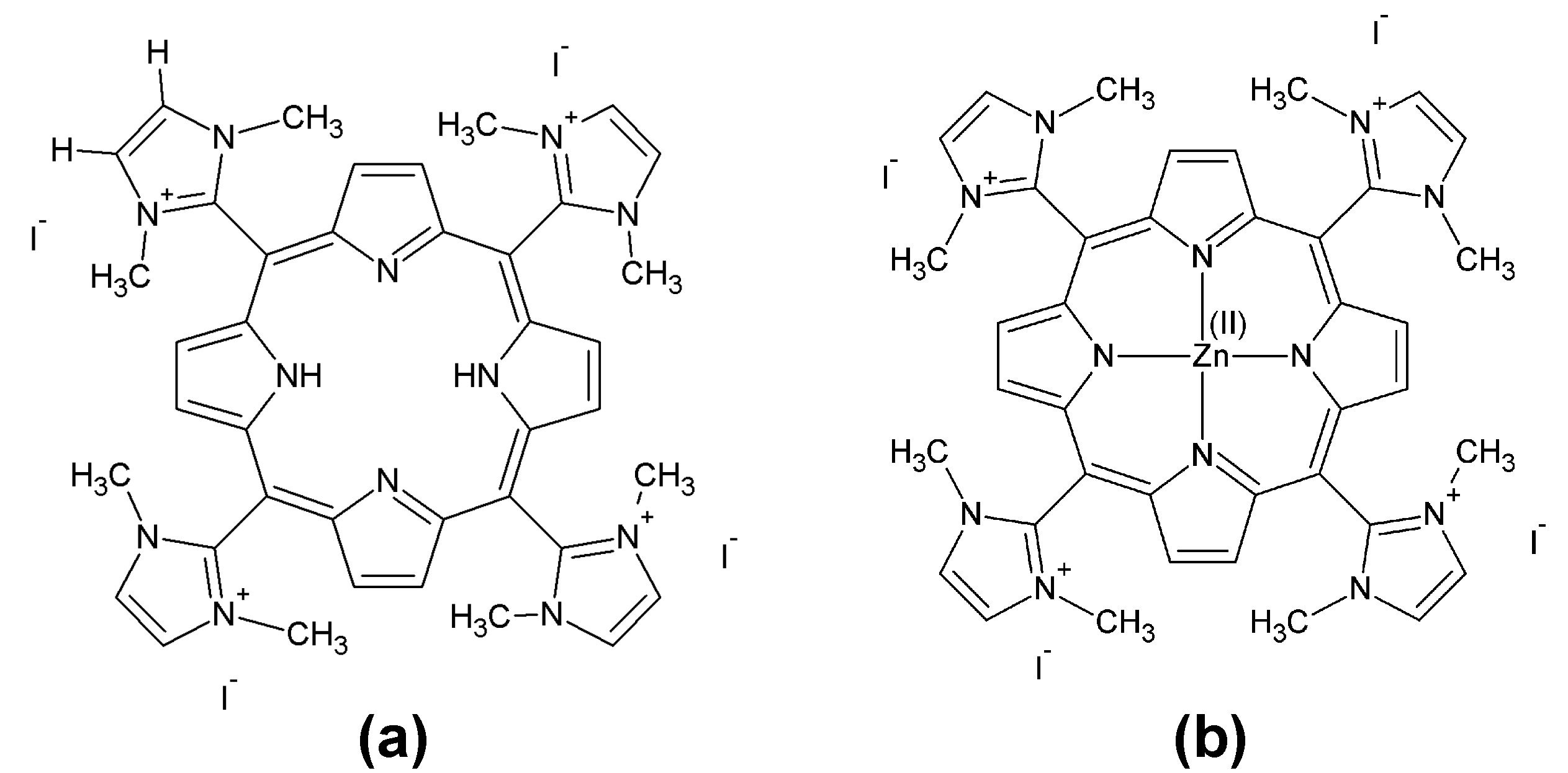
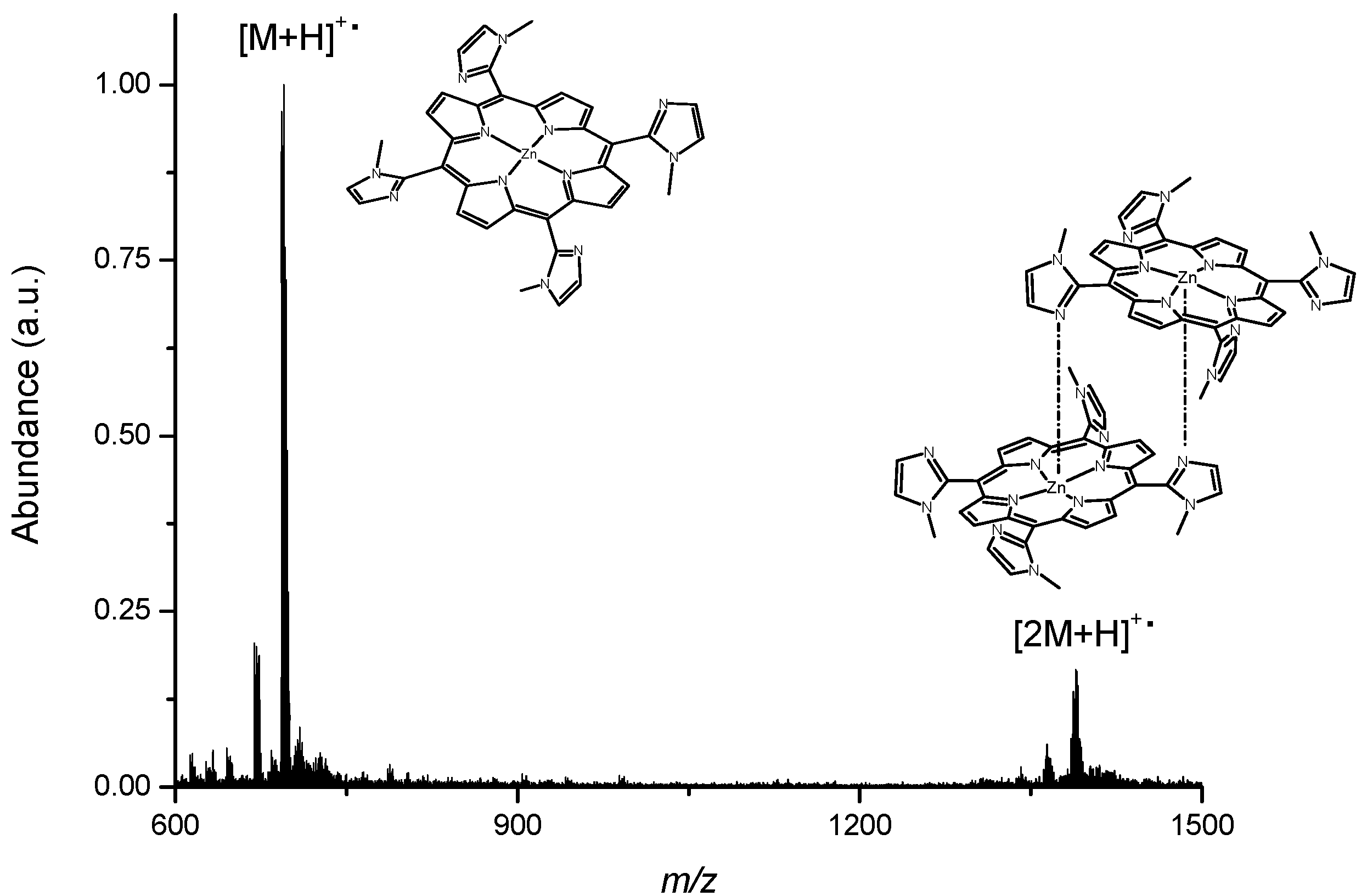
2.1. Synthesis and Characterization

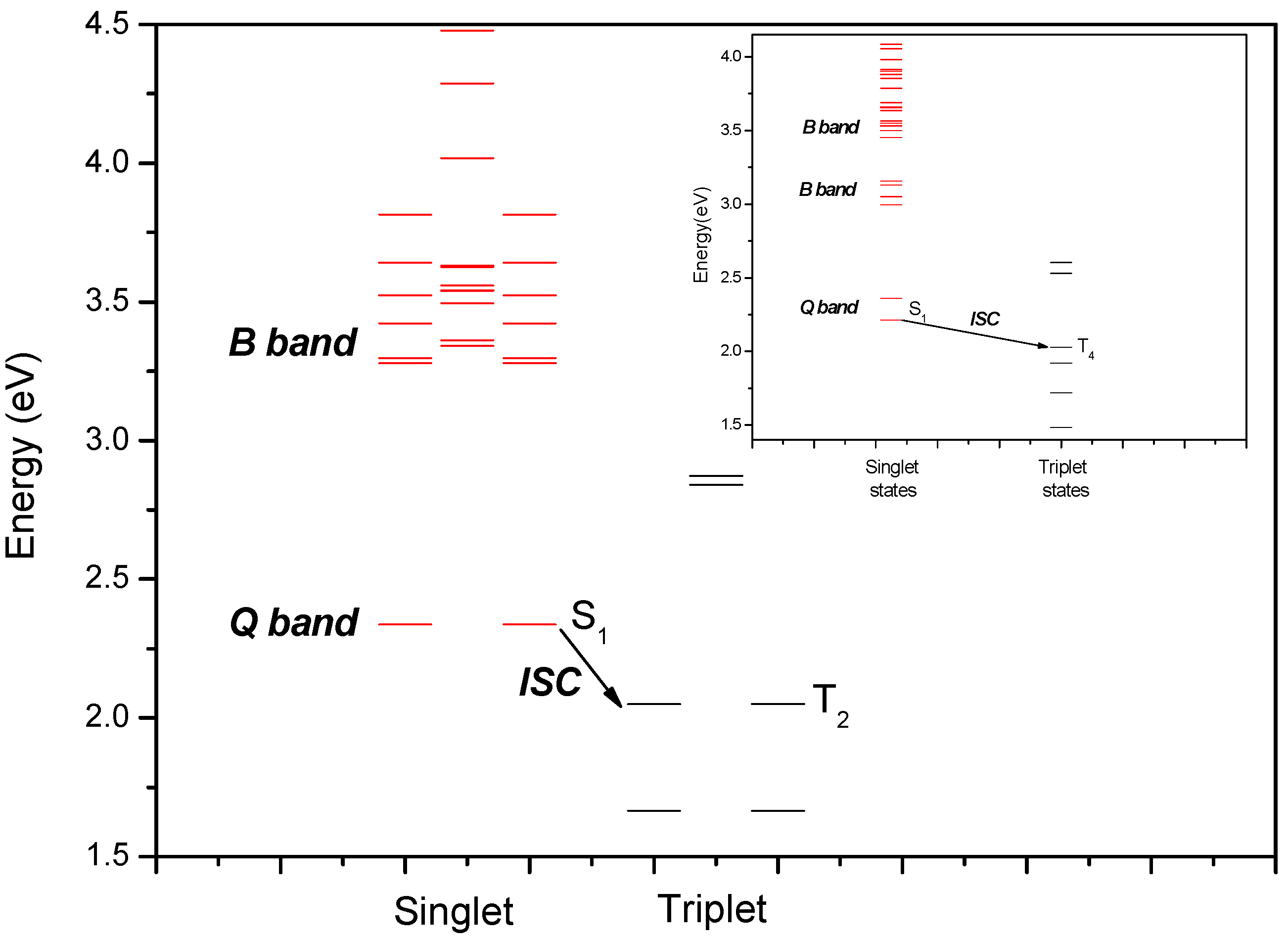
2.2. Spectroscopic Measurements and Quantum Chemical Studies
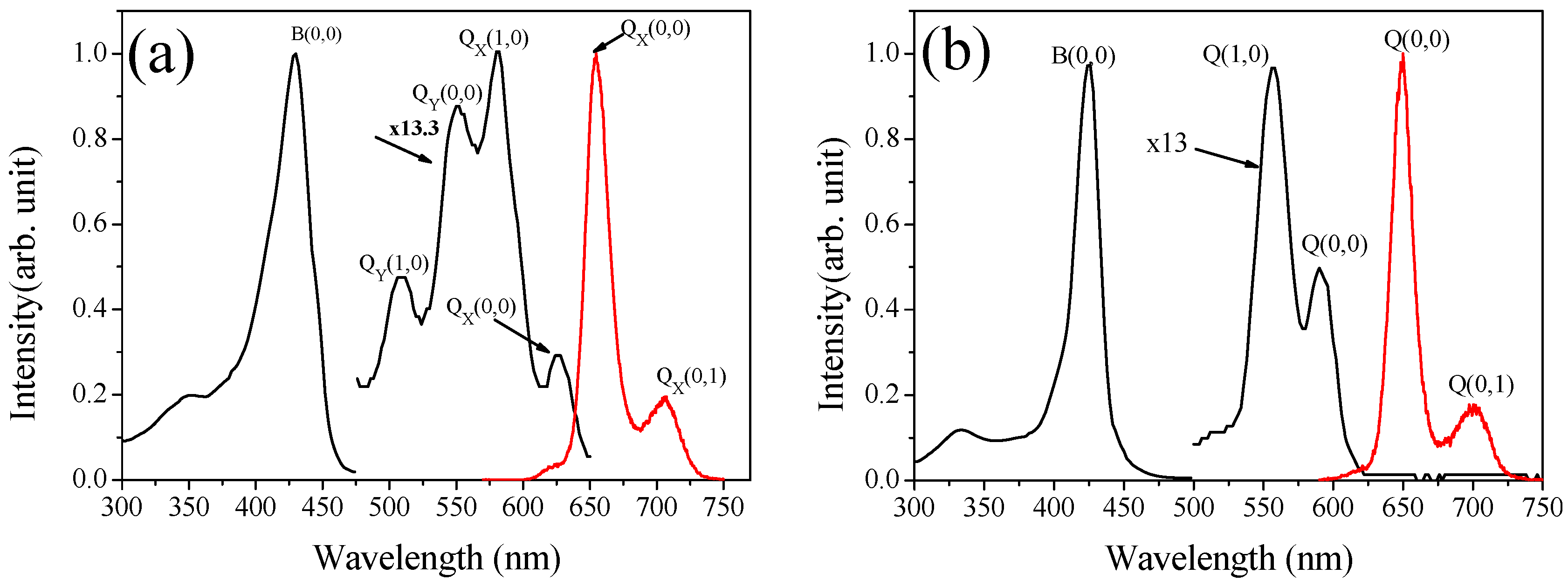
2.2.1. Absorption spectrum


{kind=link}
{kind=link}
{kind=link}
{kind=link}
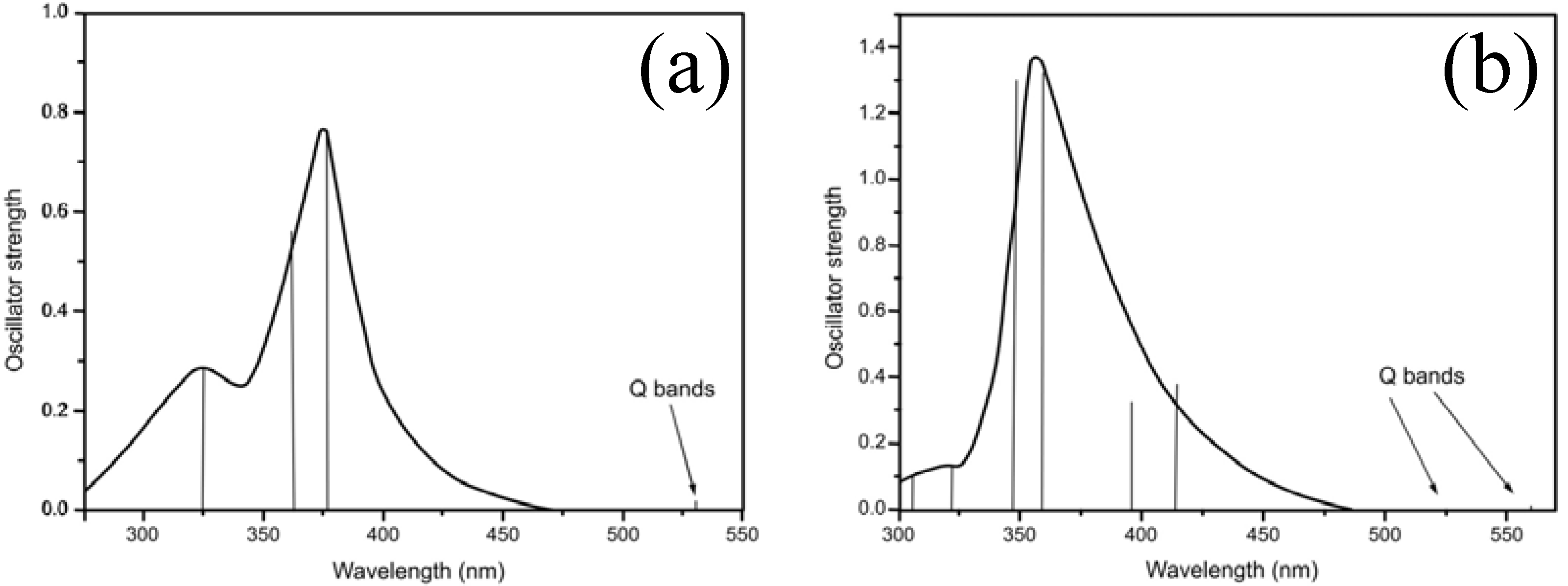{kind=link}
{kind=link}
| Solvent | λmax (Soret) (nm) | λmax (Q band)(nm) | |||
|---|---|---|---|---|---|
| Qy(1,0) | Qy(0,0) | Qx(1,0) | Qx(0,0) | ||
| Dimethylformamide | 435 | 546 | 559 | 585 | 625 |
| Dimethylsulfoxide | 417 | 511 | 544 | 581 | 633 |
| Acetonitrile | 429 | 507 | 561 | 595 | 632 |
| 2-Propanol | 431 | 508 | 554 | 582 | 625 |
| 1-Propanol | 433 | 507 | 554 | 583 | 625 |
| Ethanol | 429 (359) | 509 | 551 | 581 (525) | 627 (560) |
| Methanol | 426 | 507 | 548 | 581 | 631 |
| Ethylene glycol | 412 | 508 | 543 | 581 | 634 |
| Water | 406 | 506 | 540 | 578 | 630 |
| Solvent | λmax (Soret) (nm) | λmax (Q band) (nm) | |
|---|---|---|---|
| Q(1,0) | Q(0,0) | ||
| Dimethylformamide | 432 | 561 | 596 |
| Dimethylsulfoxide | 425 | 555 | 590 |
| Acetonitrile | 430 | 561 | 596 |
| 2-Propanol | 427 | 560 | 593 |
| 1-Propanol | 428 | 559 | 593 |
| Ethanol | 425 (376) | 557 | 591 (530) |
| Methanol | 422 | 555 | 589 |
| Ethylene glycol | 424 | 554 | 589 |
| Water | 417 | 551 | 586 |


2.2.2. Fluorescence spectrum
| Solvent | ΦF | ET(30)/kcal mol−1 | ||
|---|---|---|---|---|
| H2-TDMImP | Zn(TDMImP) | |||
| N,N–Dimethylformamide | 0.004 | 0.001 | 43.2 | |
| Dimethylsulfoxide | 0.008 | 0.010 | 45.1 | |
| Acetonitrile | 0.003 | 0.002 | 45.6 | |
| 2-Propanol | 0.006 | 0.003 | 48.4 | |
| 1-Propanol | 0.007 | 0.004 | 50.7 | |
| Ethanol | 0.007 | 0.003 | 51.9 | |
| Methanol | 0.008 | 0.008 | 55.4 | |
| ethylene glycol | 0.006 | 0.011 | 56.3 | |
| Water | 0.0004 | 0.004 | 63.1 | |
3. Experimental
3.1. General
3.2. Synthesis and Characterization

3.3. Spectroscopic Measurements
3.4. Quantum Chemical Calculations
4. Conclusions
Acknowledgements
References and Notes
- Mathai, S.; Smith, T.A.; Ghiggino, K.P. Singlet oxygen quantum yields of potential porphyrin-based photosensitisers for photodynamic therapy. Photochem. Photobiol. Sci. 2007, 6, 995–1002. [Google Scholar]
- McDonald, A.R.; Franssen, N.; van Klink, G.P.M.; van Koten, G. ‘Click’ silica immobilisation of metallo-porphyrin complexes and their application in epoxidation catalysis. J. Organomet. Chem. 2009, 694, 2153–2162. [Google Scholar] [CrossRef]
- Yang, S.I.; Seth, J.; Strachan, J.P.; Gentemann, S.; Kim, D.; Holten, D.; Lindsey, J.S.; Bocian, D.F. Ground and excited state electronic properties of halogenated tetraarylporphyrins. Tuning the building blocks for porphyrin-based photonic devices. J. Porph. Phthaloc. 1999, 3, 117–147. [Google Scholar] [CrossRef]
- Campbell, W.M.; Jolley, K.W.; Wagner, P.; Wagner, K.; Walsh, P.J.; Gordon, K.C.; Schmidt-Mende, L.; Nazeeruddin, M.K.; Wang, Q.; Grätzel, M.; Officer, D.L. Highly Efficient Porphyrin Sensitizers for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2007, 111, 11760–11762. [Google Scholar]
- Pavinatto, F.J.; Gameiro, A.F., Jr.; Hidalgo, A.A.; Dinelli, L.R.; Romualdo, L.L.; Batista, A.A.; Barbosa Neto, N.M.; Ferreira, M.; Oliveira, O.N., Jr. Langmuir and Langmuir–Blodgett (LB) films of tetrapyridyl metalloporphyrins. Appl. Surf. Sci. 2008, 254, 5946–5952. [Google Scholar]
- Garbo, G.M.; Fingar, V.H.; Wieman, T.J.; Noakes, E.B., III.; Haydon, P.S.; Cerrito, P.B.; Kessel, D.H.; Morgan, A.R. In Vivo and In Vitro Photodynamic Studies with Benzochlorin Iminium Salts Delivered by a Lipid Emulsion. Photochem. Photobiol. 1998, 68, 561–568. [Google Scholar] [CrossRef]
- Milanesio, M.E.; Alvarez, M.G.; Silber, J.; Rivarola, V.; Durantini, E.N. Photodynamic activity of monocationic and non-charged methoxyphenylporphyrin derivatives in homogeneous and biological media. Photochem. Photobiol. Sci. 2003, 2, 926–933. [Google Scholar]
- Kee, H.L.; Bhaumik, J.; Diers, J.J.R.; Mroz, P.; Hamblin, M.R.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Photophysical characterization of imidazolium-substituted Pd(II), In(III), and Zn(II) porphyrins as photosensitizers for photodynamic therapy. J. Photochem. Photobiol. A 2008, 200, 346–355. [Google Scholar] [CrossRef]
- Milgrom, L.R.; Dempsey, P.J.F.; Yahioglu, G. 5,10,15,20-tetrakis(N-protected-imidazol-2-yl)porphyrins. Tetrahedron 1996, 52, 9877–9890. [Google Scholar]
- Rose, E.; Cardonpilotaz, A.; Quelquejeu, M.; Bernard, N.; Kossanyi, A.; Desmazieres, B. Efficient Preparation of the α,α,β,β-Atropoisomer of meso-Tetrakis(0-aminopheny1)porphyrin. J. Org. Chem. 1995, 60, 3919–3920. [Google Scholar] [CrossRef]
- Kobuke, Y.; Miyaji, H. Supramolecular Organization of Imidazolyl-Porphyrin to a Slipped Cofacial Dimer. J. Am. Chem. Soc. 1994, 116, 4111–4112. [Google Scholar] [CrossRef]
- Nagata, N.; Kugimiya, S.; Kobuke, Y. Antenna functions of 5,15-bis(imidazol-4-yl)-10,20-bis(4-dodecyloxyphenyl)-porphyrin supramolecular assembly through imidazole–imidazole hydrogen bonding. Chem. Comm. 2000, 1389–1390. [Google Scholar]
- Kobuke, Y. Artificial Light-Harvesting Systems by Use of Metal Coordination. Eur. J. Inorg. Chem. 2006, 2006, 2333–2351. [Google Scholar] [CrossRef]
- Bonifassi, P.; Ray, P.C.; Leszczynski, J. Effect of central metal ions on first hyperpolarizability of unsymmetrical metal porphyrins. Chem. Phys. Lett. 2006, 431, 321–325. [Google Scholar]
- De Paula, R.; Simões, M.M.Q.; Neves, M.G.P.M.S.; Cavaleiro, J.A.S. Homogeneous olefin epoxidation catalysed by an imidazolium-based manganese porphyrin. Catal. Comm. 2008, 10, 57–60. [Google Scholar] [CrossRef]
- Tjahjono, D.H.; Akutsu, T.; Yoshioka, N.; Inoue, H. Cationic porphyrins bearing diazolium rings: Synthesis and their interaction with calf thymus DNA. Biochim. Biophys. Acta 1999, 1472, 333–343. [Google Scholar] [CrossRef]
- Day, B.J. Catalytic antioxidants: A radical approach to new therapeutics. Drug Disc. Today 2004, 9, 557–566. [Google Scholar] [CrossRef]
- Crapo, J.D.; Michael, B.J.D.; Trova, P.; Gauuan, P.J.F.; Kitchen, D.B.; Fridovich, I.; Batinic-Haberle, I. U.S. Patent 2003.
- Montanari, F.; Casella, L. Metalloporphyrin Catalyzed Oxidations; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994; p. 17. [Google Scholar]
- Kaufmann, T.; Shamsai, B.; Lu, R.S.; Bau, R.; Miskelly, G.M. Separation of the Rotational Isomers of Tetrakis(N-Methyl-2-pyridiniumyl)porphyrin and the Crystal Structure of α,α,α,β-(Tetrakis(N-methyl-2pyridiniumyl)porphyrin)copperHexacyanoferrate. Inorg. Chem. 1995, 34, 5073–5079. [Google Scholar] [CrossRef]
- Drexler, C.; Hosseini, M.W.; Planeix, J.M.; Stupka, G.; De Cian, A.; Fischer, J. Design, synthesis and structural studies on polynucleating ligands based on atropoisomerism of catechol bearing porphyrins. Chem. Comm. 1998, 689–690. [Google Scholar]
- Satake, A.; Shoji, O.; Kobuke, Y. Supramolecular array of imizazolylethynyl-zinc-porphyrin. J. Organomet. Chem. 2007, 692, 635–644. [Google Scholar] [CrossRef]
- Kachadourian, R.; Srinivasan, N.; Haney, C.A.; Stevens, R.D. An LDI-TOF and ESI mass spectrometry study of a series of β-substituted cationic metalloporphyrins. J. Porph. Phthaloc. 2001, 5, 507–511. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: San Diego, CA, USA, 1992. [Google Scholar]
- George, R.G.; Padmanabhan, M. Solvent effects on some new meso-aryl substituted octabromoporphyrins. Proc. Ind. Acad. Sci. 2003, 115, 263–271. [Google Scholar] [CrossRef]
- Yu, H.Z.; Baskin, J.S.; Zewail, A.H. Ultrafast Dynamics of Porphyrins in the Condensed Phase: II. Zinc Tetraphenylporphyrin. J. Phys. Chem. A 2002, 106, 9845–9854. [Google Scholar] [CrossRef]
- Liu, X.; Yeow, E.K.L.; Velate, S.; Steer, R.P. Photophysics and spectroscopy of the higher electronic states of zinc metalloporphyrins: A theoretical and experimental study. Phys. Chem. Chem. Phys. 2006, 8, 1298–1309. [Google Scholar]
- Nguyen, K.A.; Pachter, R. Ground state electronic structures and spectra of zinc complexes of porphyrin, tetraazaporphyrin, tetrabenzoporphyrin, and phthalocyanine: A density functional theory study. J. Chem. Phys. 2001, 114, 10757–10767. [Google Scholar] [CrossRef]
- Nguyen, K.A.; Day, P.N.; Pachter, R.; Tretiak, S.; Chernyak, V.; Mukamel, S. Analysis of Absorption Spectra of Zinc Porphyrin, Zinc meso-Tetraphenylporphyrin, and Halogenated Derivatives. J. Phys. Chem. A 2002, 106, 10285–10293. [Google Scholar]
- Spellane, P.J.; Gouterman, M.; Antipas, A.; Kim, S.; Liu, Y.C. Porphyrins.40. Electronic-spectra and 4-orbital energies of frees-base, zinc, copper, and palladium tetrakis(perfluorophenyl) porphyrins. Inorg. Chem. 1980, 19, 386–391. [Google Scholar] [CrossRef]
- Caricato, M.; Ingrosso, F.; Mennucci, B.; Tomasi, J. A time-dependent polarizable continuum model: Theory and application. J. Chem. Phys. 2005, 122, 154501–154510. [Google Scholar] [CrossRef]
- Santoro, F.; Barone, V.; Gustavsson, T.; Improta, R. Solvent Effect on the Singlet Excited-State Lifetimes of Nucleic Acid Bases: A Computational Study of 5-Fluorouracil and Uracil in Acetonitrile and Water. J. Am. Chem. Soc. 2006, 128, 16312–16322. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpete, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. Extensive TD-DFT investigation of the first electronic transition in substituted azobenzenes. Chem. Phys. Lett. 2008, 465, 226–229. [Google Scholar] [CrossRef]
- Kappe, C.O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Loupy, A. Microwaves in Organic Synthesis, 2nd ed; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- De Paula, R.; Faustino, M.A.F.; Pinto, D.C.G.A.; Neves, M.G.P.M.S; Cavaleiro, J.A.S. Kinetic study of meso-tetraphenylporphyrin synthesis under microwave irradiation. J. Heter. Chem. 2008, 45, 453–459. [Google Scholar]
- De Paula, R. Novos Derivados Porfirínicos: Síntese e Avaliação de Propriedades Catalíticas. PhD. Thesis, University of Aveiro, Aveiro, Portugal, 2009. [Google Scholar]
- Kappe, C.; Dallinger, D. Controlled microwave heating in modern organic synthesis: Highlights from the 2004-2008 literature. Mol. Divers. 2009, 13, 71–193. [Google Scholar] [CrossRef]
- de Souza, R.O.M.A.; Antunes, O.A.C.; Kroutil, W.; Kappe, C.O. Kinetic Resolution of rac-1-Phenylethanol with Immobilized Lipases: A Critical Comparison of Microwave and Conventional Heating Protocols. J. Org. Chem. 2009, 74, 6157–6162. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1999; p. 52. [Google Scholar]
- Chang, T.L.; Cheung, H.C. Solvent effects on the photoisomerization rates of the zwitterionic and the cationic forms of rhodamine B in protic solvents. J. Phys. Chem. 1992, 96, 4874–4878. [Google Scholar] [CrossRef]
- Gonçalves, P.J.; Aggarwal, L.P.F.; Marquezin, C.A.; Ito, A.S.; De Boni, L.; Barbosa Neto, N.M.; Rodrigues, J.J., Jr.; Zílio, S.C.; Borissevitch, I.E. Effects of interaction with CTAB micelles on photophysical characteristics of meso-tetrakis(sulfonatophenyl) porphyrin. J. Photochem. Photobiol. A 2006, 181, 378–384. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03 (Revision E.01), Gaussian Inc.: Wallingford, CT, USA, 2004.
- Sample Availability: Samples of the compounds are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Machado, A.E.H.; Gomes, W.R.; Araújo, D.M.S.; Miglio, H.S.; Ueno, L.T.; Paula, R.D.; Cavaleiro, J.A.S.; Neto, N.M.B. Synthesis and Spectroscopic Characterization of Two Tetrasubstituted Cationic Porphyrin Derivatives. Molecules 2011, 16, 5807-5821. https://doi.org/10.3390/molecules16075807
Machado AEH, Gomes WR, Araújo DMS, Miglio HS, Ueno LT, Paula RD, Cavaleiro JAS, Neto NMB. Synthesis and Spectroscopic Characterization of Two Tetrasubstituted Cationic Porphyrin Derivatives. Molecules. 2011; 16(7):5807-5821. https://doi.org/10.3390/molecules16075807
Chicago/Turabian StyleMachado, Antonio E.H., Weverson R. Gomes, Diesley M.S. Araújo, Hércules S. Miglio, Leonardo T. Ueno, Rodrigo De Paula, José A.S. Cavaleiro, and Newton M. Barbosa Neto. 2011. "Synthesis and Spectroscopic Characterization of Two Tetrasubstituted Cationic Porphyrin Derivatives" Molecules 16, no. 7: 5807-5821. https://doi.org/10.3390/molecules16075807
APA StyleMachado, A. E. H., Gomes, W. R., Araújo, D. M. S., Miglio, H. S., Ueno, L. T., Paula, R. D., Cavaleiro, J. A. S., & Neto, N. M. B. (2011). Synthesis and Spectroscopic Characterization of Two Tetrasubstituted Cationic Porphyrin Derivatives. Molecules, 16(7), 5807-5821. https://doi.org/10.3390/molecules16075807