Ruthenium-Catalyzed Selective Hydrogenation of bis-Arylidene Tetramic Acids. Application to the Synthesis of Novel Structurally Diverse Pyrrolidine-2,4-diones


 ,
,
Abstract
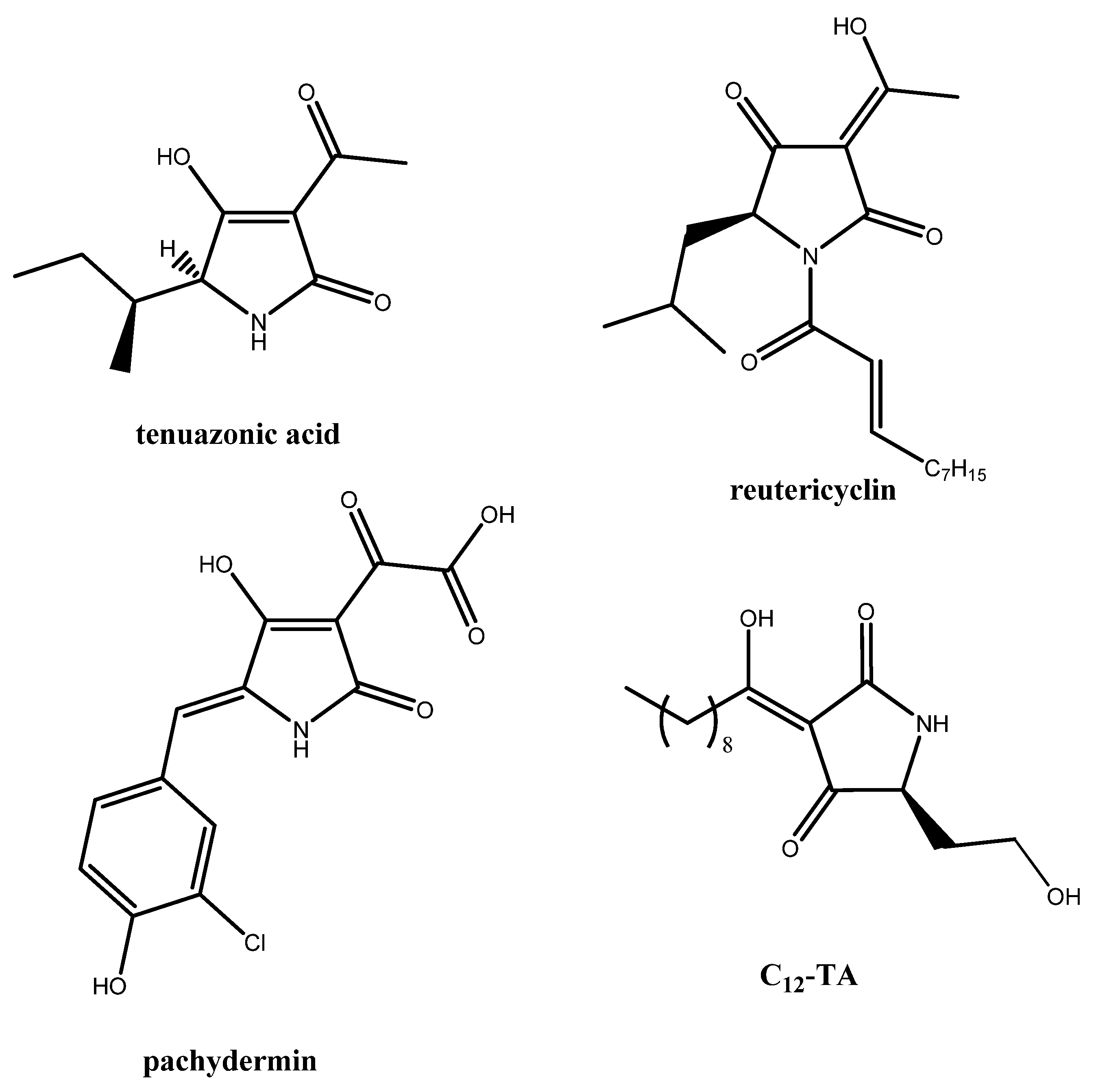
:1. Introduction

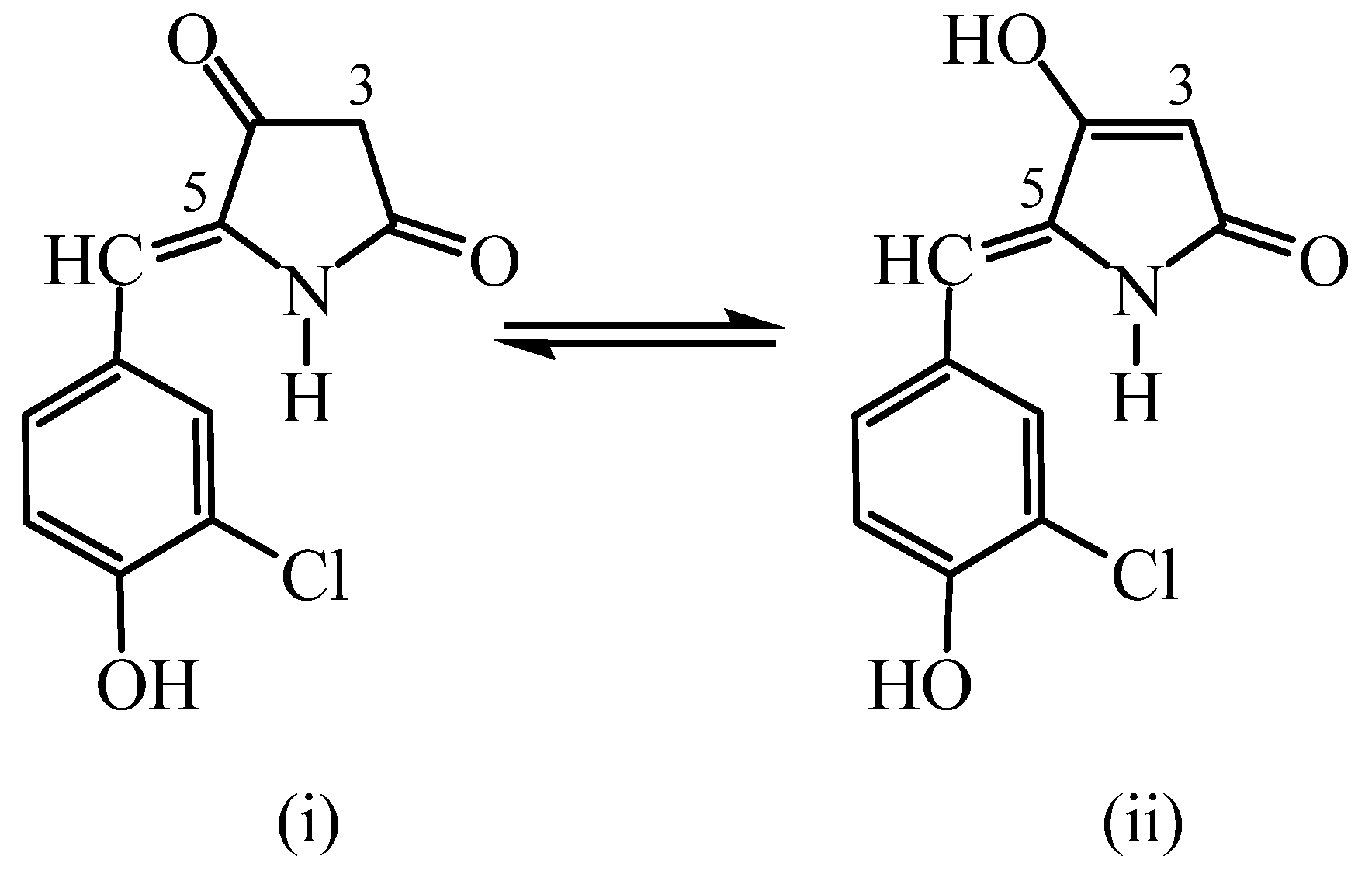
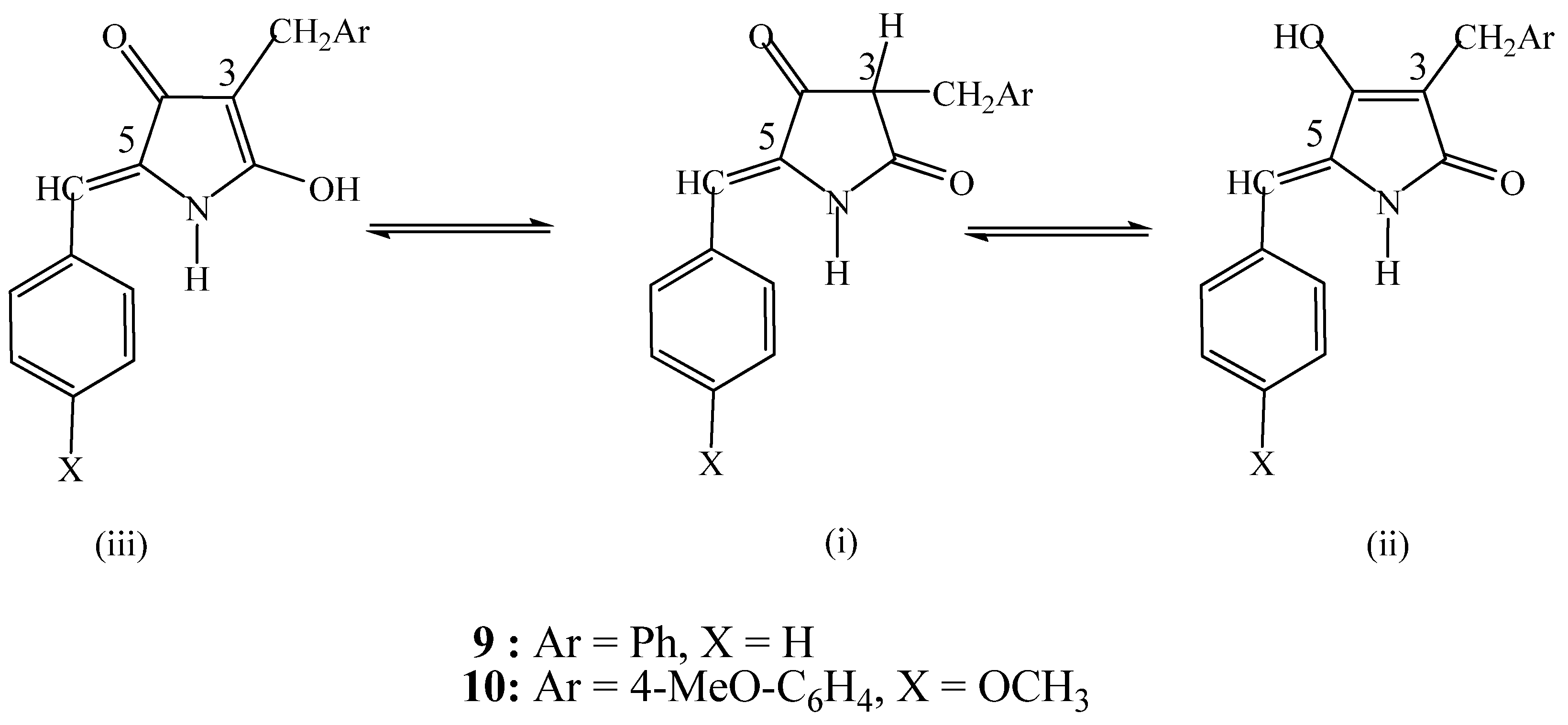
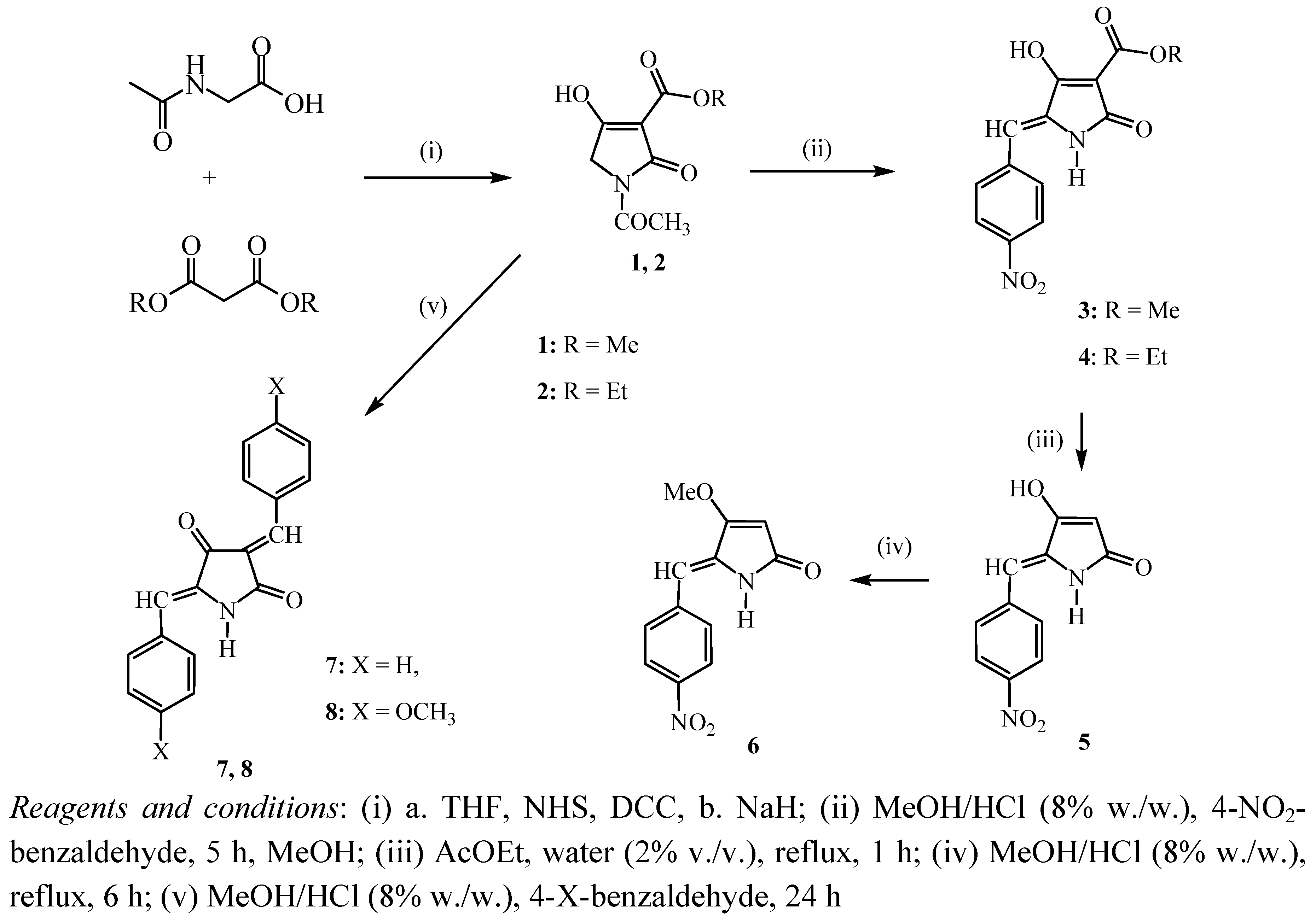
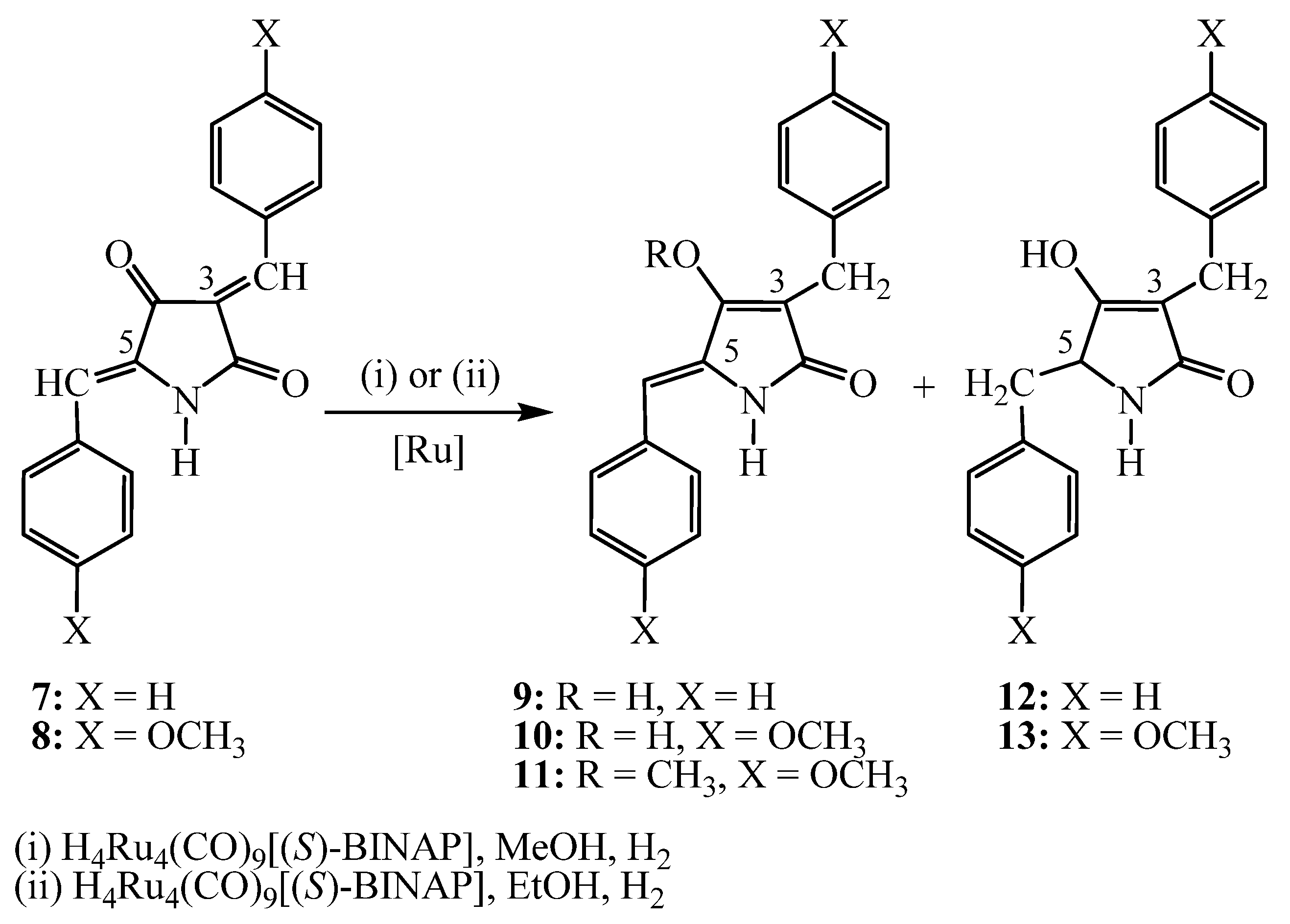
2. Results and Discussion



- (1). Polar solvents (methanol, ethanol) and
- (2). Non-polar solvents (tetrahydrofuran, dichloromethane)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | S/C | Solvent/DCM | Temp | Press | Time | Conversion% a | |
|---|---|---|---|---|---|---|---|
| (°C) | (bar) | (h) | 9 | 12 | |||
| 1 | 250 | MeOH (24:1) | 100 | 60 | 20 | 4.7 | 94.2 |
| 2 | 500 | EtOH (24:1) | 100 | 60 | 19 | 94.0 | 5.9 |
| 3 | 250 | MeOH (24:1) | 60 | 60 | 20 | 78.8 | 21.1 |
| 4 | 493 | THF (20:1) | 100 | 60 | 20 | 94.4 | 5.5 |
| Entry | S/C | Solvent/DCM | Temp | Press | Time | Conversion% a | |
|---|---|---|---|---|---|---|---|
| (°C) | (bar) | (h) | 10 | 13 | |||
| 5 | 120 | MeOH (24:1) | 100 | 60 | 20 | 24.8 | 73.9 |
| 6 | 405 | MeOH (24:1) | 100 | 60 | 20 | 44.0 | 51.6 |
| 7 b | 405 | MeOH (24:1) | 100 | 60 | 41 | 28.8 | 47.5 |
| 8 | 429 | MeOH (24:1) | 80 | 60 | 20 | 13.8 | 84.4 |
| 9 | 429 | MeOH (24:1) | 60 | 60 | 20 | 93.3 | 2.9 |
| 10 | 227 | MeOH (24:1) | 40 | 60 | 20 | 86.2 | 3.7 |
| 11 | 405 | MeOH (24:1) | 80 | 40 | 20 | 34.0 | 64.7 |
| 12 | 405 | MeOH (24:1) | 80 | 20 | 20 | 53.2 | 45.7 |
| 13 | 405 | EtOH (24:1) | 80 | 60 | 20 | 95.0 | 5.0 |
| 14 | 700 | THF (20:1) | 100 | 60 | 20 | 25.4 | 8.9 |


3. Experimental
3.1. Preparation of solvents and substrates
3.2. Synthesis of 5-arylidenetetramic acids
3.3. Procedure for the hydrogenation reaction
3.4. Synthesis of hydrogenation products
4. Conclusions
Acknowledgments
Conflict of Interest
References and Notes
- Schobert, R.; Schlenk, A. Tetramic and Tetronic Acids: An Update on New Derivatives and Biological Aspects. Bioorg. Med. Chem. 2008, 16, 4203–4221. [Google Scholar] [CrossRef]
- Gitterman, C.O. Antitumor, Cytotoxic, and Antibacterial Activities of Tenuazonic Acid and Congeneric Tetraniic Acids. J. Med. Chem. 1965, 8, 483–486. [Google Scholar] [CrossRef]
- Aoki, S.; Higuchi, K.; Ye, Y.; Satari, R.; Kobayashi, M. Melophlins A and B, Novel Tetramic Acids Reversing the Phenotype of ras-Transformed Cells, from the Marine Sponge Melophlus sarassinorum. Tetrahedron 2000, 56, 1833–1836. [Google Scholar]
- Höltzel, A.; Gänzle, M.G.; Nicholson, G.J.; Hammes, W.P.; Jung, G. The First Low Molecular Weight Antibiotic from Lactic Acid Bacteria: Reutericyclin, a New Tetramic Acid. Angew. Chem. Int. Ed. 2000, 39, 2666–2768. [Google Scholar]
- Lang, G.; Cole, A.L.J.; Blunt, J.W.; Munro, M.H.G. An Unusual Oxalylated Tetramic Acid from the New Zealand Basidiomycete Chamonixia pachydermis. J. Nat. Prod. 2006, 69, 151–153. [Google Scholar] [CrossRef]
- Lou, L.; Qian, G.; Xie, Y.; Hang, J.; Chen, H.; Zaleta-Rivera, K.; Li, Y.; Shen, Y.; Dussault, P.H.; Liu, F.; Lou, L.D. Biosynthesis of HSAF, a Tetramic Acid-Containing Macrolactam from Lysobacter enzymogenes. J. Am. Chem. Soc. 2011, 133, 643–645. [Google Scholar]
- Kaufmann, G.F.; Sartorio, R.; Lee, S.; Rogers, C.J.; Meijer, M.M.; Moss, J.A.; Clapham, B.; Brogan, A.P.; Dickerson, T.J.; Janda, K.D. Revisiting Quorum Sensing: Discovery of Additional Chemical and Biological Functions for 3-oxo-N-acylhomoserine Lactones. Proc. Natl. Acad. Sci. USA 2005, 102, 309–314. [Google Scholar]
- Xu, W.; Cai, X.; Jung, M.E.; Tang, Y. Analysis of Intact and Dissected Fungal Polyketide Synthase-Nonribosomal Peptide Synthetase in vitro and in Saccharomyces cerevisiae. J. Am. Chem. Soc. 2010, 132, 13604–13607. [Google Scholar]
- Mawer, I.M.; Kulagowski, J.J.; Leeson, P.D.; Grimwood, S.; Marshall, G.R. Tetramic acids as novel glycine site antagonists. Bioorg. Med. Chem. Lett. 1995, 22, 2643–2648. [Google Scholar]
- Matiadis, D.; Igglessi-Markopoulou, O. Design and Synthesis of Optically Active Esters of γ-Amino-β-oxo Acids as Precursors for the Synthesis of Tetramic Acids Derived from L-Serine, L-Tyrosine, and L-Threonine. Eur. J. Org. Chem. 2010, 5989–5995. [Google Scholar]
- Prousis, K.C.; Markopoulos, J.; Mckee, V.; Igglessi-Markopoulou, O. Efficient construction of functionalized 5-carboxymethyl tetramic acids using N-Ac-L-aspartic anhydride as chiral building block. Tetrahedron 2010, 66, 3944–3950. [Google Scholar] [CrossRef]
- Heaton, B.T. Mechanisms in Homogeneous Catalysis: A Spectroscopic Approach; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Ikariya, T.; Murata, K.; Noyori, R. Bifunctional transition metal-based molecular catalysts for asymmetric syntheses. Org. Biomol. Chem. 2006, 4, 393–406. [Google Scholar] [CrossRef]
- Noyori, R.; Ohkuma, T. Asymmetric Catalysis by Architectural and Functional Molecular Engineering: Practical Chemo- and Stereoselective Hydrogenation of Ketones. Angew. Chem. Int. Ed. 2001, 40, 40–73. [Google Scholar] [CrossRef]
- Shang, G.; Li, W.; Zhang, X. Catalytic Asymmetric Synthesis, 3rd; Ojima, I., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2010; pp. 343–436, Chapter 7. [Google Scholar]
- Tang, W.; Zhang, X. New Chiral Phosphorus Ligands for Enantioselective Hydrogenation. Chem. Rev. 2003, 103, 3029–3070. [Google Scholar] [CrossRef]
- Morris, R.H. Handbook of Homogeneous Hydrogenation; de Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Volume 1, pp. 45–70, Chapter 3. [Google Scholar]
- Smit, C.; Fraaije, M.W.; Minnaard, A.J. Reduction of Carbon−Carbon Double Bonds Using Organocatalytically Generated Diimide. J. Org. Chem. 2008, 73, 9482–9485. [Google Scholar] [CrossRef]
- Han, A.; Jiang, X.; Civiello, R.L.; Degnan, A.P.; Chaturvedula, P.V.; Macor, J.E.; Dubowchik, G.M. Catalytic Asymmetric Syntheses of α-Amino and α-Hydroxyl Acid Derivatives. J. Org. Chem. 2009, 74, 3993–3996. [Google Scholar]
- Li, X.; Li, L.; Tang, Y.; Zhong, L.; Cun, L.; Zhu, J.; Liao, J.; Deng, J. Chemoselective Conjugate Reduction of α,β-Unsaturated Ketones Catalyzed by Rhodium Amido Complexes in Aqueous Media. J. Org. Chem. 2010, 75, 2981–2988. [Google Scholar] [CrossRef]
- Kosal, A.D.; Ashfeld, B.L. Titanocene-catalyzed conjugate reduction of alpha,beta-unsaturated carbonyl derivatives. Org. Lett. 2010, 12, 44–47. [Google Scholar] [CrossRef]
- Llamas, T.; Arrayás, R.G.; Carretero, J.C. Catalytic Asymmetric Conjugate Reduction of β,β-Disubstituted α,β-Unsaturated Sulfones. Angew. Chem. Int. Ed. 2007, 46, 3329–3332. [Google Scholar] [CrossRef]
- Ngai, M.Y.; Kong, J.R.; Krische, M.J. Hydrogen-Mediated C−C Bond Formation: A Broad New Concept in Catalytic C−C Coupling. J. Org. Chem. 2007, 1063–1072. [Google Scholar]
- Genet, J.P. Modern Reduction Methods; Andersson, P.G., Munslow, I.J., Eds.; Wiley VCH: Weinheim, Germany, 2008; pp. 1–38, Chapter 1. [Google Scholar]
- Dobbs, D.A.; Vanhessche, K.P.; Brazi, E.; Rautenstrauch, V.V.; Lenoir, J.Y.; Genêt, J.P.; Wiles, J.; Bergens, S.H. Industrial Synthesis of (+)-cis-Methyl Dihydrojasmonate by Enantioselective Catalytic Hydrogenation; Identification of the Precatalyst. Angew. Chem. Int. Ed. 2000, 39, 1992–1995. [Google Scholar] [CrossRef]
- Athanasellis, G.; Gavrielatos, E.; Igglessi-Markopoulou, O. One-Pot Synthesis of Optically Active Tetramic Acids from Amino Acids Mediated by 1-Hydroxybenzotriazole. Synlett 2001, 10, 1653–1655. [Google Scholar]
- Athanasellis, G.; Gavrielatos, E.; Igglessi-Markopoulou, O. Synthesis and spectroscopic studies of 5-arylidene-3-substituted tetramic acids as possible substrates for catalytic asymmetric hydrogenation. J. Heterocycl. Chem. 2001, 38, 1203–1208. [Google Scholar]
- Poschenrieder, H.; Höfner, G.; Stachel, H.-D. 5-Arylidene-3-aryl-pyrrolidine-2,4-diones with affinity to the N-methyl-D-aspartate (glycine site) receptor, Part I. Arch. Pharm. Pharm. Med. Chem. 1998, 331, 389–394. [Google Scholar] [CrossRef]
- Nishi, T.; Kitamura, M.; Ohkuma, T.; Noyori, R. Synthesis of statine and its analogues by homogeneous asymmetric hydrogenation. Tetrahedron Lett. 1988, 29, 6327–6330. [Google Scholar]
- Kitamura, M.; Hsiao, Yi; Ohta, M.; Tsukamoto, M.; Ohta, T.; Takaya, H.; Noyori, R. General Asymmetric Synthesis of Isoquinoline Alkaloids. Enantioselective Hydrogenation of Enamides Catalyzed by BINAP- Ruthenium (II) Complexes. J. Org. Chem. 1994, 59, 297–310. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Tunik, S.P.; Pilyugina, T.S.; Koshevoy, I.O.; Selivanov, S.I.; Haukka, M.; Pakkanen, T.A. Reaction of (S)-BINAP with H4Ru4(CO)12. The First Example of Face-Bridging BINAP Coordination and 100% Stereoselectivity in Formation of a Chiral Tetranuclear Cluster Framework. Organometallics 2004, 568–579. [Google Scholar]
- Kuroki, Y.; Asada, D.; Sakamaki, Y.; Iseki, K. Synthesis of optically active partly gem-difluorinated allylic alcohols via [2,3]-Wittig rearrangements and lipase-catalyzed reaction. Tetrahedron Lett. 2000, 41, 4603–4607. [Google Scholar] [CrossRef]
- Lu, W.J.; Chen, Y.W.; Hoo, X.L. Iridium-Catalyzed Highly Enantioselective Hydrogenation of the CC Bond of α, β-Unsaturated Ketones. Angew. Chem. Int. Ed. 2008, 47, 10133–10136. [Google Scholar] [CrossRef]
- Petroliagi, M.; Igglessi-Markopoulou, O. An efficient synthesis of novel N-acetyl-3-alkanoyl and 3-dienoyl tetramic acids. J. Chem. Soc. Perkin Trans. 1 1997, 23, 3543–3548. [Google Scholar]
- Petroliagi, M.; Igglessi-Markopoulou, O. Synthesis and enantiomeric excess measurements of optically active N-acetyl tetramic acids. Tetrahedron Asymmetry 1999, 10, 1873–1875. [Google Scholar]
- Poschenrieder, H.; Stachel, H.-D.; Eckl, E.; Jax, S.; Polborn, K.; Mayer, P. Functionalized Imides by Regioselective Ozonation. Helv. Chim. Acta 2006, 89, 971–982. [Google Scholar]
- Galeotti, N.; Poncet, J.; Chiche, L.; Jouin, P. Diastereofacial Selectivity in Reduction of Chiral Tetramic Acids. J. Org. Chem. 1993, 58, 5370–5376. [Google Scholar] [CrossRef]
- Sorokina, I.K.; Alekseeva, L.M.; Parshin, V.A.; Granik, V.G. Functional derivatives of tetramic acid: New approaches to the synthesis of heterotricyclic compounds. Pharm. Chem. J. 2007, 41, 543–548. [Google Scholar] [CrossRef]
- Stachel, H.D.; Poschenrieder, H.; Burghard, H. Synthesis of 5-alkylidene-3-pyrrolin-2-ones. Z. Naturforsch. 1986, 41b, 640–644. [Google Scholar]
- Sample Availability: Contact the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karaiskos, C.S.; Matiadis, D.; Markopoulos, J.; Igglessi-Markopoulou, O. Ruthenium-Catalyzed Selective Hydrogenation of bis-Arylidene Tetramic Acids. Application to the Synthesis of Novel Structurally Diverse Pyrrolidine-2,4-diones. Molecules 2011, 16, 6116-6128. https://doi.org/10.3390/molecules16076116
Karaiskos CS, Matiadis D, Markopoulos J, Igglessi-Markopoulou O. Ruthenium-Catalyzed Selective Hydrogenation of bis-Arylidene Tetramic Acids. Application to the Synthesis of Novel Structurally Diverse Pyrrolidine-2,4-diones. Molecules. 2011; 16(7):6116-6128. https://doi.org/10.3390/molecules16076116
Chicago/Turabian StyleKaraiskos, Christos S., Dimitris Matiadis, John Markopoulos, and Olga Igglessi-Markopoulou. 2011. "Ruthenium-Catalyzed Selective Hydrogenation of bis-Arylidene Tetramic Acids. Application to the Synthesis of Novel Structurally Diverse Pyrrolidine-2,4-diones" Molecules 16, no. 7: 6116-6128. https://doi.org/10.3390/molecules16076116
APA StyleKaraiskos, C. S., Matiadis, D., Markopoulos, J., & Igglessi-Markopoulou, O. (2011). Ruthenium-Catalyzed Selective Hydrogenation of bis-Arylidene Tetramic Acids. Application to the Synthesis of Novel Structurally Diverse Pyrrolidine-2,4-diones. Molecules, 16(7), 6116-6128. https://doi.org/10.3390/molecules16076116