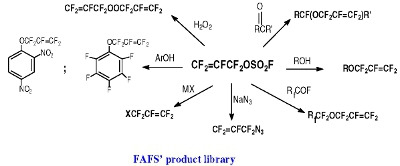
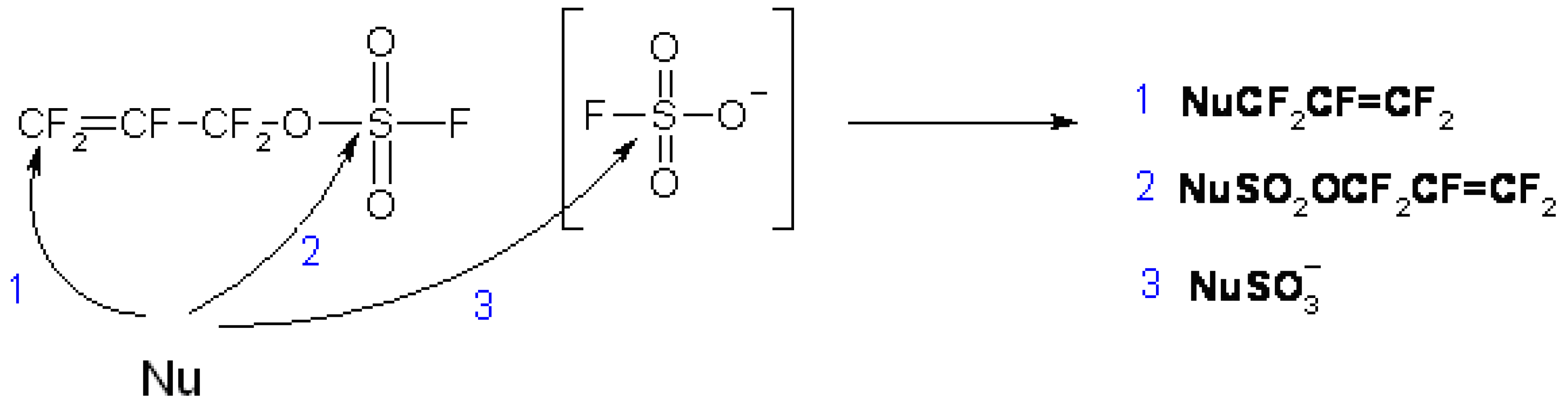
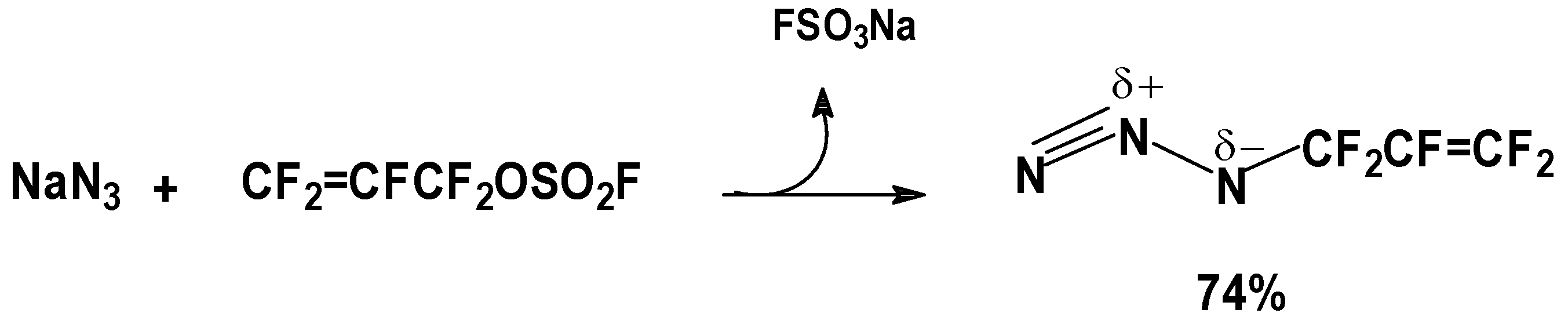
FAFS is an asymmetrical olefin and therefore it will have two centers of attack about the CF2=CF– bond: The C-3 terminal olefin carbon or the C-2 internal carbon. Furthermore, FAFS also embodies two distinct electrophilic centers: The terminal olefin and the electrophilic sulfur atom as well. These electronic features give FAFS a variety of different regiochemistries depending on the nature of the reaction.
2.2.2. Nucleophilic reactions
It will be shown that FAFS, due to its electrophilic nature, is quite reactive towards a number of different nucleophiles, including for example alcohols, yielding the corresponding fluorinated allyl ethers. Unlike what was previously reported in the literature [
4], it is subject to nucleophilic substitution by alcohols both without basic catalysis (
i.e., directly with the protonated alcohol) as well as with the corresponding conjugate base. Employing an excess of an alcohol in the presence of FAFS one always obtains the corresponding allyl ether.
Table 4 shows the selectivities and product distributions of some typical hydrogenated and partially fluorinated alcohols both with (Na
+ as the cation) and without basic catalysis.
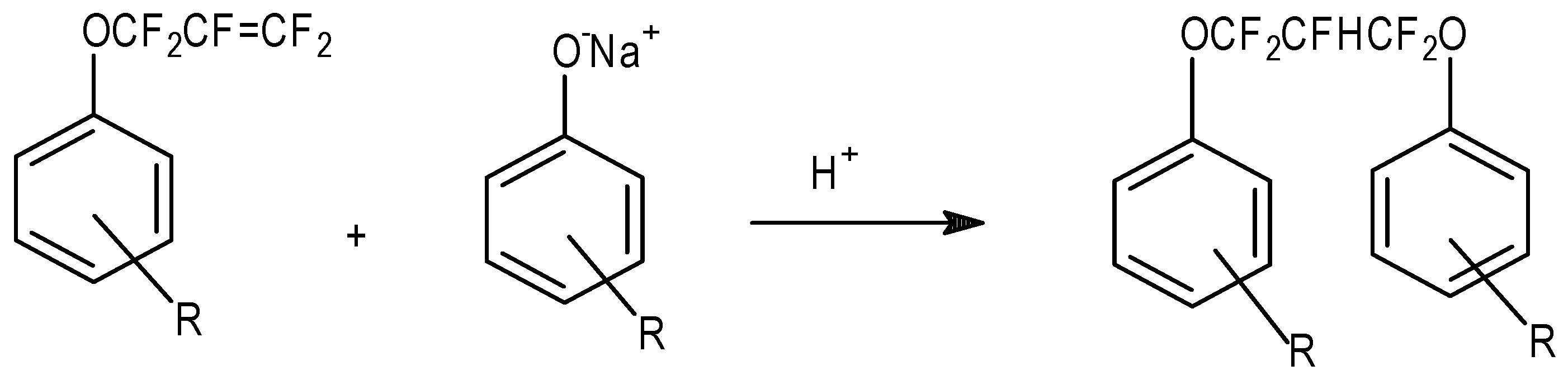
Of course, with the base catalyzed nucleophilic addition to FAFS, one must employ stoichiometric quantities of the alcohol in order to avoid a second addition of the alcoholate to the allyl ether yielding, from a general alcohol ROH, RO–CF2CFHCF2–OR. The proton in the fluorinated propyl chain comes from the solvent (generally CH3CN or glymes) employed.
Table 4.
FAFS regioselectivity with several oxygen nucleophiles.
Table 4.
FAFS regioselectivity with several oxygen nucleophiles.
| Trial | Nucleophile | Conv. (FAFS) | Products (selectivity %) |
|---|
| 14 | CH3OH | 100% | CH3OCF2CF=CF2 (100%) |
| 15 | CH3ONa | 100% | CH3OCF2CF=CF2 (100%) |
| 16 | C6H5OH | 54% | C6H5OCF2CF=CF2 (86%)/CF2=CFCF2OSO2OC6H5 (14%) |
| 17 | C6H5ONa | 98% | C6H5OCF2CF=CF2 (87%)/CF2=CFCF2OSO2OC6H5 (13%) |
| 18 | CF3CH2OH | 46% | CF3CH2OCF2CF=CF2 (85%)/CF2=CFCF2OSO2OCH2CF3 (15%) |
| 19 | CF3CH2ONa | 97% | CF3CH2OCF2CF=CF2 (86%)/CF2=CFCF2OSO2OCH2CF3 (14%) |
| 20 | C6H5CH2OH | 96% | C6H5CH2OCF2CF=CF2 (95%)/CF2=CFCF2OSO2OCH2C6H5 (5%) |
| 21 | C6H5CH2ONa | 99% | C6H5CH2OCF2CF=CF2 (94%)/CF2=CFCF2OSO2OCH2C6H5 (6%) |
| 22 | C6F5OH | 30% | C6F5OCF2CF=CF2 (86%)/CF2=CFCF2OSO2OC6F5 (14%) |
| 23 | C6F5ONa | 100% | C6F5OCF2CF=CF2 (90%)/CF2=CFCF2OSO2OC6F5 (10%) |
Table 4 shows that there are at least two distinct regiochemistries involved in the nucleophilic addition to FAFS: One yields an allyl ether (main product) and the other a sulfate ester (minor product).
Scheme 4 shows the three possible sites of attack of a general nucleophile to FAFS. Taking reaction 1 depicted in
Scheme 4 into consideration, unlike the hypofluorite radical addition shown in
Scheme 3, nucleophilic attack was almost exclusively (>98.5/1.5) observed on the terminal olefin yielding a secondary anion. Pathway 1 is an Addition/Elimination (A/E) mechanism of the nucleophile to FAFS’ terminal double bond. The main driving force of the reaction is the powerful leaving group FSO
3−. Furthermore, it is known from the literature that attack by a nucleophile on the sp
3 carbon in highly fluorinated molecules does not occur [
2]. On the other hand, Pathway 2 is a Substitution (S
N) reaction by the nucleophile on FAFS’ sulfur atom yielding a sulfate ester. The driving force of this reaction is the electropositive sulfur and the relatively good leaving group, F
−. In very few instances and with particularly acidic fluoro alcohols, Pathway 3 was also observed: Once FSO
3M (M = H, Metal) is formed by Pathway 1, a second nucleophile can attack FSO
3M’s electropositive sulfur atom, displace F
− and form the general product NuOSO
2M.
As can be seen from
Table 5 there is a direct correlation between the alcohol’s pK
a and the A/E
vs. S
N product distribution shown in
Table 4.
Scheme 4.
Different modes of nucleophilic attack of a general nucleophile Nu: on FAFS.
Scheme 4.
Different modes of nucleophilic attack of a general nucleophile Nu: on FAFS.
Table 5.
Correlation between an oxygen nucleophile’s pKa and substitution selectivity on FAFS.
Table 5.
Correlation between an oxygen nucleophile’s pKa and substitution selectivity on FAFS.
| ROH | pKa | CF2=CFCF2OSO2OR % |
|---|
| C6H5OH | 9,9 | 14 |
| CF3CH2OH | 12,4 | 15 |
| C6H5CH2OH | 15 | 5 |
| CH3OH | 16 | 0 |
Alcohols with a pKa less than 13 and therefore relatively “acidic” either by resonance effect (as in phenol, pKa = 9.9) or by inductive effect (as in trifluoroethanol, pKa = 12.4) give a higher percentage of SN product (Pathway 2). On the other hand, methanol (pKa = 16) and benzyl alcohol (pKa = 15), which are more basic, give almost exclusively the A/E product (Pathway 1).
Another interesting feature that emerges from the data presented in
Table 4 is that the A/E
vs. S
N selectivity remains practically unchanged regardless to whether the nucleophile is a charged species (oxyanion) or a species with a free unpaired electron doublet on the oxygen atom (alcohol). This leads us to assert that the regioselectivity observed is determined not only by the particular electronic nature of the nucleophile (pK
a due to resonance or inductive effects, oxyanion
vs. protonated alcohol) but also on the electronic nature of FAFS’s terminal olefin
vs. FAFS’s sulfur atom.
Finally, regardless of the regiochemistry observed, the base catalyzed addition of an alcohol to FAFS is a much faster reaction as evidenced by the higher conversions of FAFS with the conjugate base
vs. the free alcohol. The striking differences in regioselectivities observed thus far, led us to investigate if there was a “cation” effect on regioselectivity as well. It is known in the literature that the electronic nature of the nucleophiles is not only governed by inductive and mesomeric effects, but also by the Hard-Soft-Acid-Base theory of Lewis [
12] and Pearson [
13] whose trends are shown in
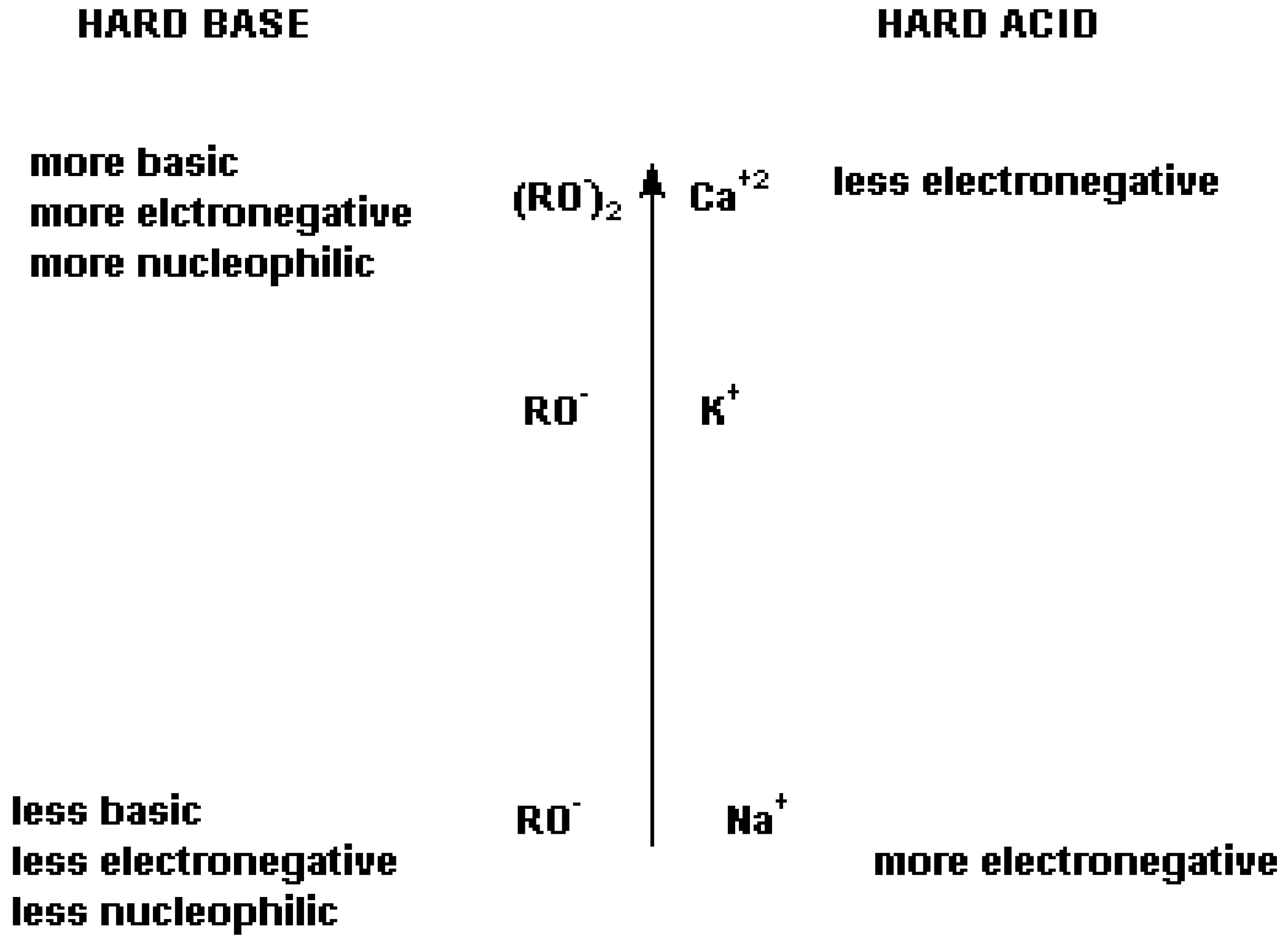
Figure 2.
Figure 2.
Lewis [
12] and Pearson’s [
13] representation of the “Hard-Soft-Acid-Base theory concerning anions and cations.
Figure 2.
Lewis [
12] and Pearson’s [
13] representation of the “Hard-Soft-Acid-Base theory concerning anions and cations.
It becomes clear that, based on the regiochemistry considerations made thus far, varying the nucleophile’s cation may vary the regiochemistry for the nucleophilic attack on FAFS.
We therefore used pentafluorophenol (pK
a = 8.9) [
14] as a model compound to study the effects of Ca
2+, K
+ and Na
+ cations on regiochemistry. The averaged results of the cation effect on regiochemistry are shown in
Table 6 along with the
19F-NMR details shown in
Figure 3a–c. All reactions were performed in anhydrous THF with a stoichiometric quantity of nucleophiles with respect to the moles of FAFS.
Table 6.
Addition/Elimination vs. Substitution molar selectivities on FAFS as a function of the cation.
Table 6.
Addition/Elimination vs. Substitution molar selectivities on FAFS as a function of the cation.
| Trial | Nucleophile | TR(°C) | C6F5OCF2CF=CF2/C6F5O–SO2–OCF2CF=CF2 |
|---|
| 24 | (C6F5O)2Ca | 40 °C | 13/87 |
| 25 | C6F5OK | 40 °C | 60/40 |
| 26 | C6F5ONa | 40 °C | 90/10 |
Figure 3.
19F-NMR (200 MHz) spectrum of the Addition/Elimination (“Allyl”) vs. Substitution (“Ester”) products after reaction between FAFS and (a) C6F5ONa, (b) C6F5OK and (c) (C6F5O)2Ca.
Figure 3.
19F-NMR (200 MHz) spectrum of the Addition/Elimination (“Allyl”) vs. Substitution (“Ester”) products after reaction between FAFS and (a) C6F5ONa, (b) C6F5OK and (c) (C6F5O)2Ca.
The experimental data reported in
Table 6 confirm the HSAB theory summarized in
Figure 3: In going from a Na
+ cation to a Ca
2+ cation the hard base alcoholate becomes progressively more ionically charged or, in other words, less covalently bound, and therefore more susceptible to attacking FAFS’ very electropositive sulfur atom. Therefore, the main product of the nucleophilic addition of sodium perfluoro phenolate and FAFS is C
6F
5OCF
2CF=CF
2 (A/E selectivity = 90%), the minor product is CF
2=CFCF
2OSO
3C
6F
5 (S
N selectivity = 10%); on the other hand, the main product employing calcium phenolate is the sulfate ester (S
N selectivity = 87%), and the minor product is the corresponding perfluoro allyl ether (A/E; selectivity= 13%). Pure compounds from Trials 24 and 26 were isolated by flash silica gel chromatography and identified by GC-MS. This permitted us to unequivocally assign the
19F-NMR frequencies (in ppm) observed in
Figure 3a–c and shown in
Table 7.
Table 7.
Specific 19F-NMR (300 MHz) frequencies observed for Addition/Elimination (a–c’)vs. Substitution (d–f’) products.
Table 7.
Specific 19F-NMR (300 MHz) frequencies observed for Addition/Elimination (a–c’)vs. Substitution (d–f’) products.
| | a | b | c | c’ | d | e | f | f’ |
|---|
| –OaCF2bCF=c,c’CF2 | −70.6 | −189 | −89.8 | −103.2 | - | - | - | - |
| –OSO2O-dCF2eCF=f,f’CF2 | - | - | - | - | −71.2 | −189.5 | −94.2 | −105.8 |
Therefore, in a base catalyzed addition between an alcohol’s conjugate base and FAFS, in order to selectively obtain an A/E product, i.e., an allyl ether as the main product, the cation must be Na+.
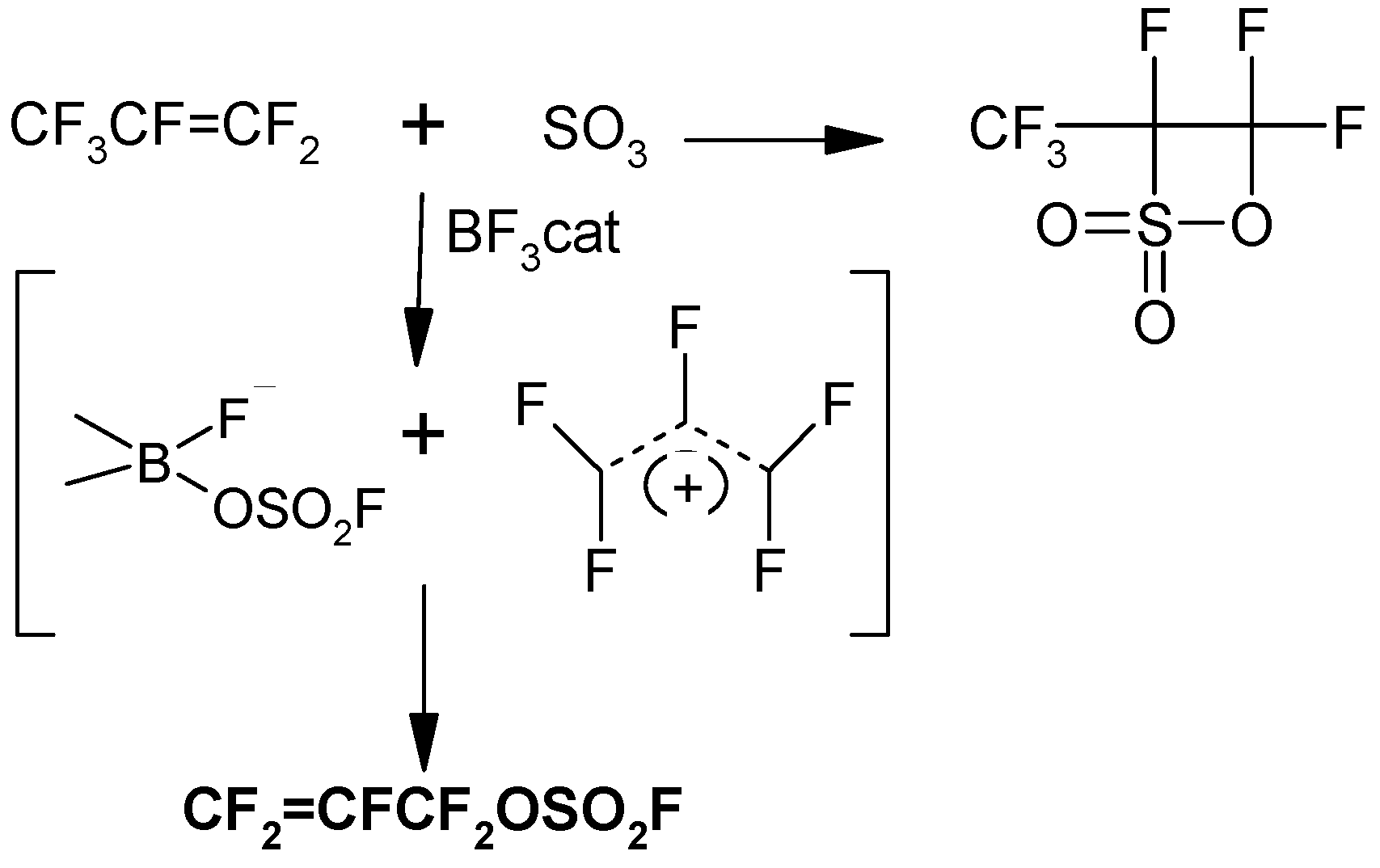
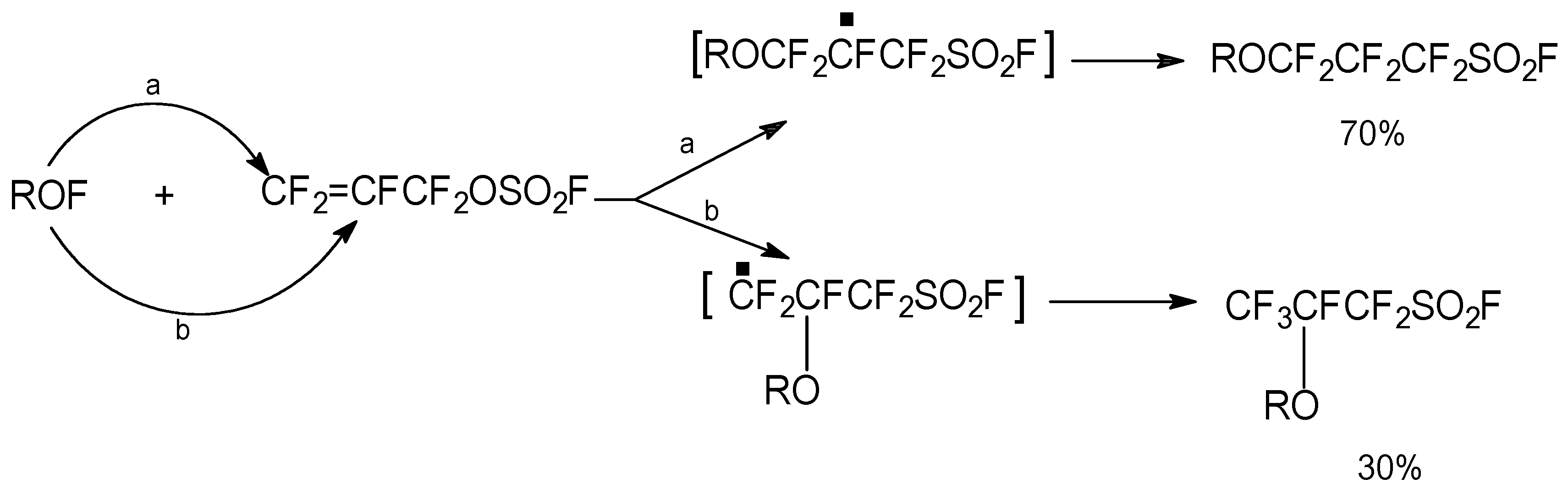
One of the few well documented A/E reactions in the literature is the sum of a metal halide MX, to FAFS where X = I, Br, Cl [
4]. It becomes immediately obvious that ICF
2CF=CF
2 is a hydrolytically stable synthon of FAFS, but with only one possible regioisomer obtainable due to the absence of the electrophilic sulfur atom. Therefore, if quantitative selectivity towards A/E is necessary, ICF
2CF=CF
2 can be synthesized
in situ (see Experimental), according to a slightly modified reaction procedure with respect to the literature [
4], and immediately added to the nucleophile according to
Scheme 5. Complete regioselectivity towards the allyl ether is obtained with isolated yields ranging from 55%–85% depending upon the alcohol.
Scheme 5.
Allyl iodide mediated synthesis of allyl ethers.
Scheme 5.
Allyl iodide mediated synthesis of allyl ethers.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















