Synthesis of cis- and trans-3-Aminocyclohexanols by Reduction of β-Enaminoketones

Abstract
:1. Introduction
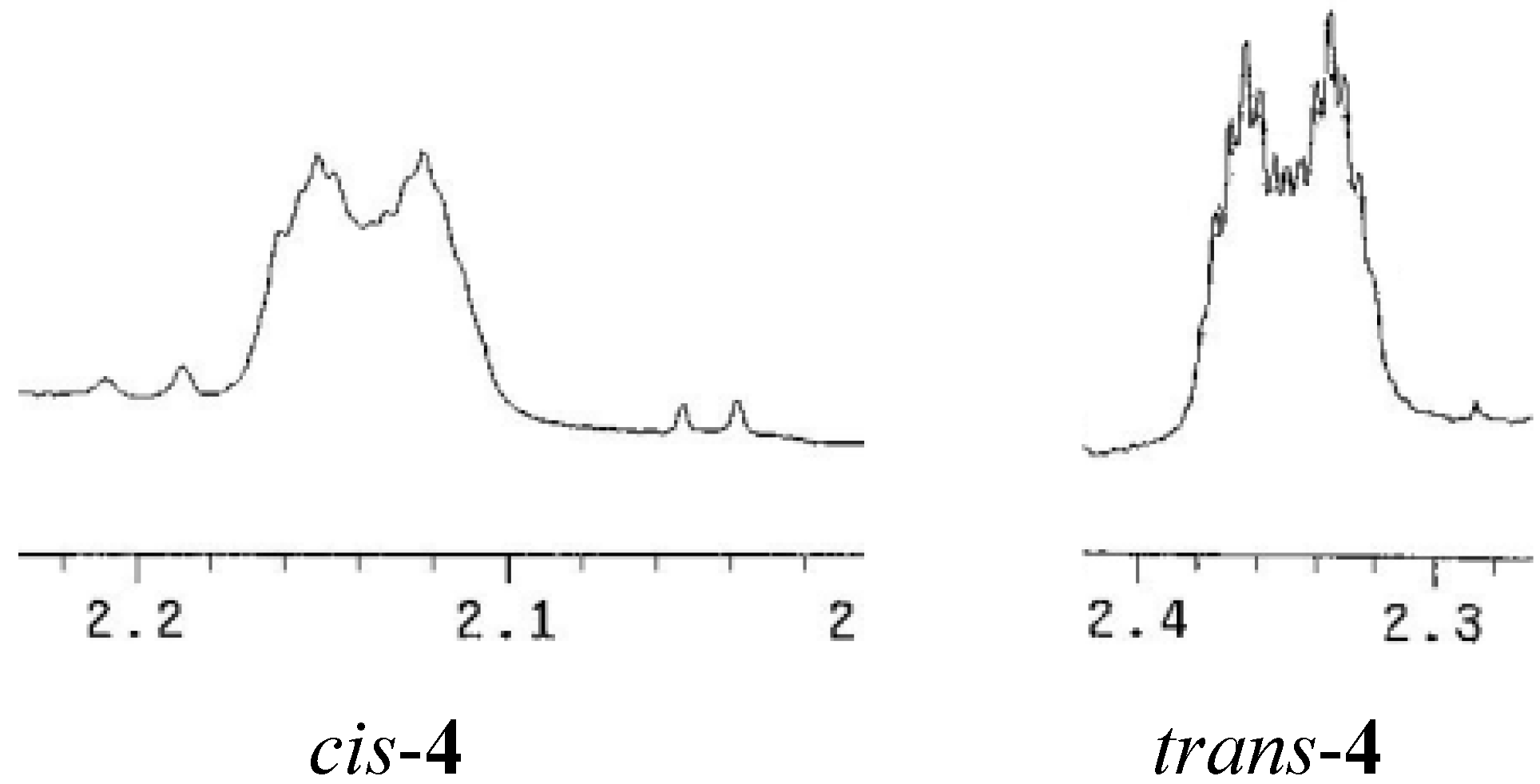
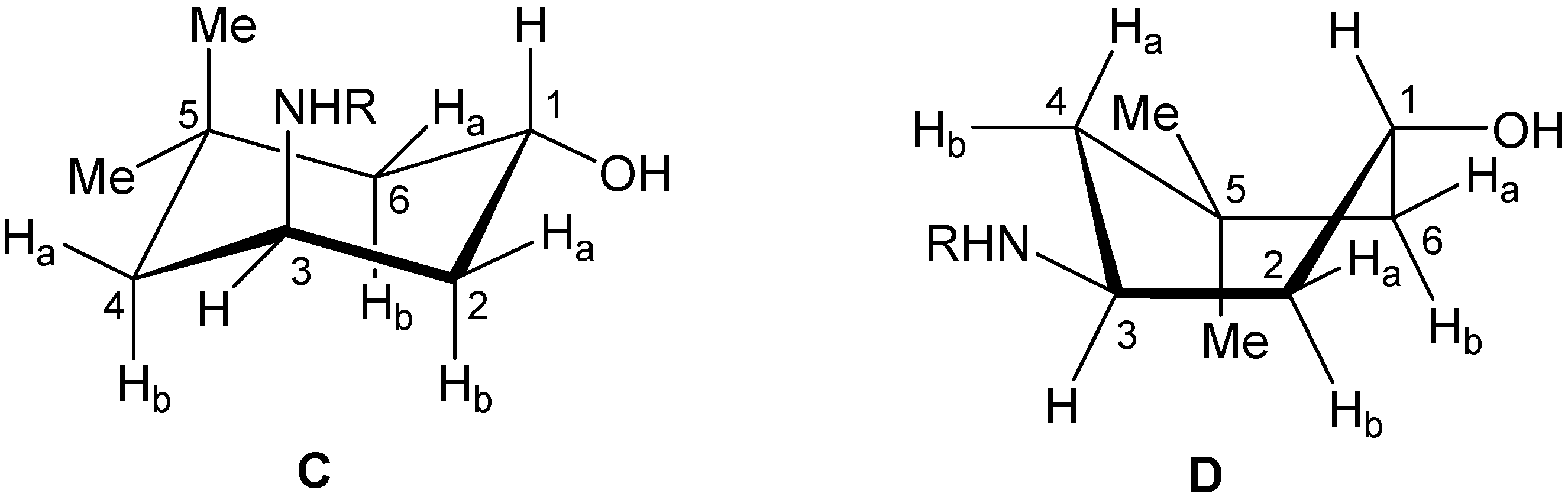
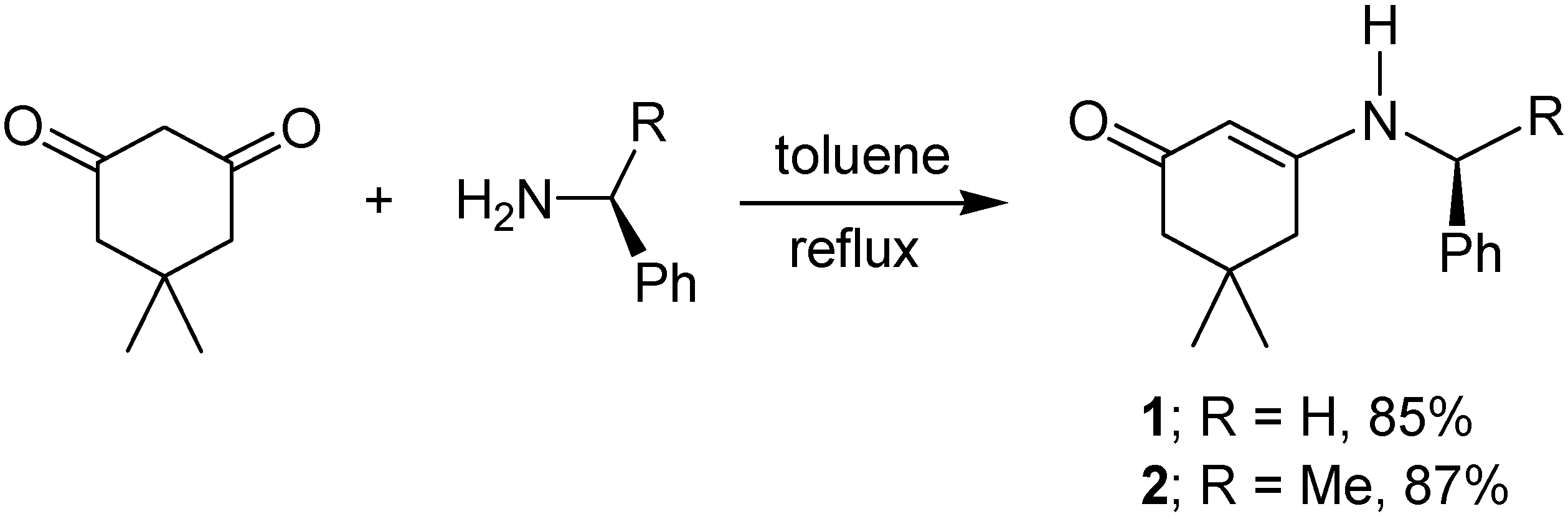
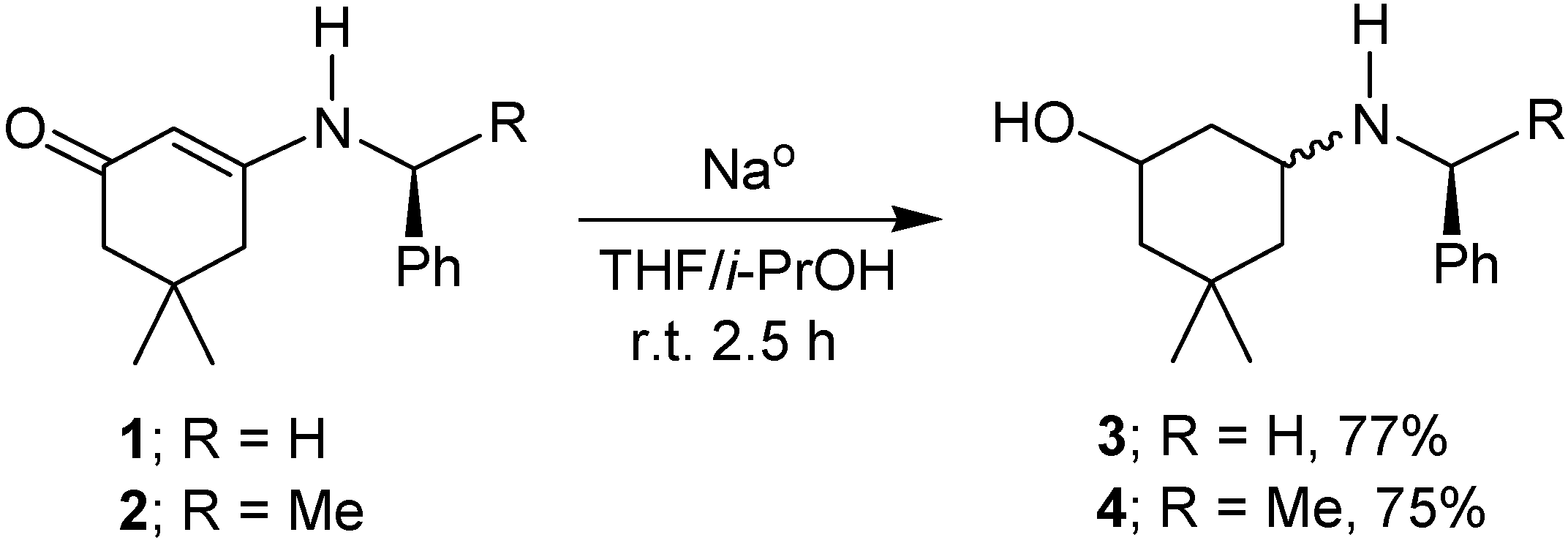
2. Results and Discussion





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| cis-4 | trans-4 | ||||
|---|---|---|---|---|---|
| Proton | 1H δ(ppm), J (Hz) | Carbon | 13C δ (ppm) | 1H δ(ppm), J (Hz) | 13C δ (ppm) |
| H1 | 3.65 (tt, J = 11.2, 4.8, 1H) | C1 | 66.8 | 3.64 (tt, J = 10.8, 4.4, 1H) | 67.1 |
| H2a | 2.13 (m, Jgem = 11.6, 1H) | C2 | 43.3 | 2.35 (dddd, J = 11.6, 5.6, 4.2, 1H) | 42.6 |
| H3 | 2.53 (tt, J = 11.6, 4.0, 1H) | C3 | 49.5 | 2.59 (tt, J = 11.6, 4.0, 1H) | 49.3 |
| H4a | 1.70 (ddt, J = 12.8, 3.6, 2.0, 1H) | C4 | 44.7 | 1.50 (m, 1H) | 46.5 |
| H5 | - - | C5 | 31.8 | - - | 31.7 |
| H6a | 1.63 (ddt, J = 12.4, 4.0, 2.0, 1H) | C6 | 48.1 | 1.63 (ddt, J = 12.4, 4.0, 2.0, 1H) | 48.4 |
| H7 | 0.97 (s, 3H) | C7 | 33.3 | 0.93 (s, 3H) | 33.2 |
| H8 | 0.70 (s, 3H) | C8 | 26.0 | 0.75 (s, 3H) | 26.2 |
| H9 | 4.00 (q, J = 6.4, 1H) | C9 | 55.1 | 4.03 (q, J = 6.8, 1H) | 54.8 |
| H10 | 1.42 (d, J = 6.4, 3H) | C10 | 24.3 | 1.40 (d, J = 6.8, 3H) | 24.9 |
| C6H5 | 7.30–7.38 (m, 5H) | Cipso | 144.3, | 7.32–7.35 (m, 5H) | 145.4, 128.7, 127.1, 126.7 |
| NH, OH | 2.37 (bs, 2H) | - - | 2.01 (bs, 2H) | - - | |





| Proton | cis-4 | trans-4 |
|---|---|---|
| 1 | 3, 2eq, 4eq, Meupfield | 2eq, 6eq |
3. Experimental
3.1. General Experimental Procedures
3.2. General Procedure for the Reduction of β-Enaminoketones 1 and 2
4. Conclusions
Acknowledgements
References and Notes
- Shibahara, S.; Kondo, S.; Maeda, K.; Umezawa, H.; Ohno, M. Total synthesis of negamycin and the antipode. J. Am. Chem. Soc. 1972, 94, 4353–4354. [Google Scholar]
- Kozikowski, A.P.; Chen, Y.-Y. Intramolecular nitrile oxide cycloaddition (INOC) reactions in the indole series. 2. Total synthesis of racemic and optically active paliclavine and 5-epi-paliclavine. J. Org. Chem. 1981, 46, 5248–5250. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Izawa, T.; Kobayashi, S.; Ohno, M. Stereocontrolled synthesis of (+)-negamycin from an acyclic homoallylamine by 1,3-asymmetric induction. J. Am. Chem. Soc. 1982, 104, 6465–6466. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Kawada, A.; Natsugari, H. Stereoselective synthesis of sperabillins and related compounds. J. Chem. Soc. Perkin Trans. 1 1991, 2435–2444. [Google Scholar]
- Knapp, S. Synthesis of complex nucleoside antibiotics. Chem. Rev. 1995, 95, 1859–1876. [Google Scholar] [CrossRef]
- Sakai, R.; Kamiya, H.; Murata, M.; Shimamoto, K. Dysiherbaine: A new neurotoxic amino acid from the micronesian marine sponge Dysidea herbacea. J. Am. Chem. Soc. 1997, 119, 4112–4116. [Google Scholar] [CrossRef]
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.-P.; et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. USA 1995, 92, 2484. [Google Scholar]
- Sham, H.L.; Zhao, C.; Li, L.; Betebenner, D.A.; Saldivar, A.; Vasavanonda, S.; Kempf, D.J.; Plattner, J.J.; Norbeck, D.W. Novel lopinavir analogues incorporating non-aromatic P-1 side chains-synthesis and structure-activity relationships. Bioorg. Med. Chem. Lett. 2002, 12, 3101–3103. [Google Scholar] [CrossRef]
- Haight, A.R.; Stuk, T.L.; Allen, M.S.; Bhagavatula, L.; Fitzgerald, M.; Hannick, S.M.; Kerdesky, F.A.J.; Menzia, J.A.; Parekh, S.I.; Robbins, T.A.; et al. Reduction of an enaminone: Synthesis of the diamino alcohol core of ritonavir. Org. Process Res. Dev. 1999, 3, 94–100. [Google Scholar] [CrossRef]
- Shi, Z.; Harrison, B.A.; Verdine, G.L. Unpredictable stereochemical preferences for Mu opioid receptor activity in an exhaustively stereodiversified library of 1,4-enediols. Org. Lett. 2003, 5, 633–636. [Google Scholar] [CrossRef]
- Kondo, S.; Shibahara, S.; Takahashi, S.; Maeda, K.; Umezawa, H.; Ohno, M. Negamycin, a novel hydrazide antibiotic. J. Am. Chem. Soc. 1971, 93, 6305–6306. [Google Scholar] [CrossRef]
- Raju, B.; Mortell, K.; Anandan, S.; O’Dowd, H.; Gao, H.; Gomez, M.; Hackbarth, C.; Wu, C.; Wang, W.; Yuan, Z.; et al. N- and C-terminal modifications of negamycin. Bioorg. Med. Chem. Lett. 2003, 13, 2413–2418. [Google Scholar]
- Naidu, S.V.; Kumar, P.A. Simple and efficient approach to 1,3-aminoalcohols: Application to the synthesis of (+)-negamicyn. Tetrahedron Lett. 2007, 48, 3793–3796. [Google Scholar] [CrossRef]
- Carlier, P.R.; Lo, M.M.-C.; Lo, P.C.-K.; Richelson, E.; Tatsumi, M.; Reynolds, I.J.; Sharma, T.A. Synthesis of a potent wide-spectrum serotonin-, norepinephrine-, dopamine-reuptake inhibitor (SNDRI) and a species-selective dopamine-reuptake inhibitor based on the gamma-amino alcohol functional group. Bioorg. Med. Chem. Lett. 1998, 8, 487–492. [Google Scholar] [CrossRef]
- Wang, X.-B.; Kodama, K.; Hirose, T.; Yang, X.-F.; Zhang, G.-Y. Chirality control in the enantioselective arylation of aromatic aldehydes catalized by cis-(1R,2S)-2-benzamidocyclo-hexanecarboxylic acid derived 1,3-aminoalcohols. Tetrahedron: Asymmetry 2010, 21, 75–80. [Google Scholar] [CrossRef]
- Geng, H.; Zhang, W.; Chen, J.; Hou, G.; Zhou, L.; Zou, Y.; Wu, W.; Zhang, X. Rhodium-catalized enantioselective and diastereoselective hydrogenation of β-ketoenamides: Efficient access to anti-1,3-amino alcohols. Angew. Chem. Int. Ed. 2009, 48, 6052–6054. [Google Scholar] [CrossRef]
- Davis, F.A.; Gaspari, P.M.; Nolt, B.M.; Xu, P. Asymmetric synthesis of acyclic 1,3-amino alcohols by reduction of N-sulfinyl β-amino ketones. Formal synthesis of (-)-pinidol and (+)-epipinidol. J. Org. Chem. 2008, 73, 9619–9626. [Google Scholar] [CrossRef]
- Menche, D.; Arikan, F.; Li, J.; Rudolph, S. Directed reductive amination β-hydroxy-ketones: Convergent assembly of the ritonavir/lopinavir core. Org. Lett. 2007, 9, 267–270. [Google Scholar] [CrossRef]
- Kochi, T.; Tang, T.P.; Ellman, J.A. Asymmetric synthesis of syn- and anti-1,3-amino alcohols. J. Am. Chem. Soc. 2002, 124, 6518–6519. [Google Scholar] [CrossRef]
- Keck, G.E.; Truong, A.P. Directed reduction of β-amino ketones to syn- or anti-1,3-amino alcohol derivatives. Org. Lett. 2002, 4, 3131–3134. [Google Scholar] [CrossRef]
- Vilaplana, M.J.; Molina, P.; Arques, A.; Andrés, C.; Pedrosa, R. Synthesis of the novel chiral 1,3-amino alcohol 8-N,N-bis(Ferrocenylmethyl)amino-menthol and its use as catalyst in the enantioselective addition of diethylzinc to aldehydes. Tetrahedron: Asymmetry 2002, 13, 5–8. [Google Scholar] [CrossRef]
- Panev, S.; Linden, A.; Dimitrov, V. Chiral aminoalcohols with a menthane skeleton as catalyst for the enantioselective addition of diethylzinc to benzaldehyde. Tetrahedron: Asymmetry 2001, 12, 1313–1321. [Google Scholar] [CrossRef]
- Andrés, C.; Duque-Soladana, J.P.; Iglesias, J.M.; Pedrosa, R. Diastereoselective 5-exo-trig radical cyclisation on N-acryloyl-tetrahydro-1,3-oxazines. A novel approach to enantiopure 3-substituted pyrrolidines. Tetrahedron Lett. 1996, 37, 9085–9086. [Google Scholar]
- Liu, D.; Gao, W.; Wang, C.; Zhang, X. Practical synthesis of practical enantiopure γ-aminoalcohols by rhodium-catalyzed asymmetric hydrogenation of β-secondary-amino ketones. Angew. Chem. Int. Ed. 2005, 44, 1687–1689. [Google Scholar] [CrossRef]
- Rice, G.T.; White, M.C. Allylic C-H amination for the preparation of syn-1,3-amino alcohol motifs. J. Am. Chem. Soc. 2009, 131, 11707–11711. [Google Scholar] [CrossRef]
- Millet, R.; Träff, A.M.; Petrus, M.L.; Bäckvall, J.-E. Enantioselective synthesis of syn and anti-1,3-aminoalcohols via β-aminoketones and subsequent reduction/dynamic kinetic asymmetric transformation. J. Am. Chem. Soc. 2010, 132, 15182–15184. [Google Scholar] [CrossRef]
- Solé, C.; Whiting, A.; Gulyás, H.; Fernández, E. Highly enantio and diastereoselective synthesis of γ-amino alcohols from α,β-unsaturated imines trough a one-pot β-boration/reduction/oxidation sequence. Adv. Synth. Catal. 2011, 353, 376–384. [Google Scholar] [CrossRef]
- Anzai, M.; Yanada, R.; Fujii, N.; Ohno, H.; Ibuka, T.; Takemoto, Y. Asymmetric synthesis of β2,3-amino acids by InI-Pd(0)-promoted metalation and addition of chiral 2-vinylaziridines. Tetrahedron 2002, 58, 5231–5239. [Google Scholar] [CrossRef]
- Raghavan, S.; Rajender, A. Stereoselective synthesis of (-)-allosedamine and (1R,3R)-HPA-12 from β-p-toluenesulfonamido-γ,δ-unsaturated sulfoxide. Tetrahedron 2004, 60, 5059–5067. [Google Scholar] [CrossRef]
- Patti, A.; Pedotti, S. Chemoenzymatic access to all four enantiopure stereoisomers of 1-ferrocenyl-1,3-butanediol. Tetrahedron: Asymmetry 2006, 17, 778–785. [Google Scholar] [CrossRef]
- Barbarotto, M.; Geist, J.; Choppin, S.; Colobert, F. SmI2-coupling reaction of chiral non-racemic α-bromo-α´-sulfinyl ketones with imines: Synthesis of enantiomerically pure 2-methyl-3-amino-1-ol moieties. Tetrahedron: Asymmetry 2009, 20, 2780–2787. [Google Scholar] [CrossRef]
- Olsson, C.; Helgesson, S.; Frejd, T. New bicyclic γ- and δ-aminoalcohols as catalyst for the asymmetric diethylzinc addition to benzaldehyde. Tetrahedron: Asymmetry 2008, 19, 1484–1493. [Google Scholar] [CrossRef]
- Levy, L.M.; de Gonzalo, G.; Gotor, V. Resolution of N-protected cis- and trans-3-aminocyclohexanols via lipase-catalyzed enantioselective acylation in organic media. Tetrahedron: Asymmetry 2004, 15, 2051–2056. [Google Scholar] [CrossRef]
- Bernardelli, P.; Bladon, M.; Lorthiois, E.; Manage, A.C.; Vergne, F.; Wrigglesworth, R. Resolution of trans-3-aminocyclohexanol. Tetrahedron: Asymmetry 2004, 15, 1451–1455. [Google Scholar] [CrossRef]
- Santos, E.; Padilla, J.; Crabbé, P. Optical properties dimedonyl derivatives of α-phenylethylamine. Can. J. Chem. 1967, 45, 2275–2277. [Google Scholar] [CrossRef]
- Dudek, G.O.; Holm, R.H. Nuclear magnetic resonance studies of keto-enol equilibria. III. α,β-unsaturated-β-ketoamines. J. Am. Chem. Soc. 1962, 84, 2691–2696. [Google Scholar] [CrossRef]
- Crystal data for C16H21NO (2), Mr = 243.34 gmol−1, 0.41 × 0.34 × 0.16 mm3, monoclinic, space group P2(1), a = 9.6513(16), b = 7.0546(11), c = 21.462(4) Å, α = 90, β = 93.259(3), γ = 90°, V = 1458.9(4) Å3, Z = 4, ρcalcd = 1.108 gcm−3, θmax=25°, 5149 independent reflections, R1 = 0.0700 for 14134 reflections with I > 2σ(I) and wR2 = 0.1455 for all data, 2 parameters. Crystallographic data for the structure reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 841749.
- Bartoli, G.; Cimarelli, C.; Palmieri, G. Convenient procedure for the reduction of β-enamino ketones: Synthesis of γ-amino alcohols and tetrahydro-1,3-oxazines. J. Chem. Soc. Perkin Trans. 1 1994, 537–543. [Google Scholar]
- San Martin, R.; Martínez de Marigorta, E.; Domínguez, E. A convenient alternative route to β-aminoketones. Tetrahedron 1994, 50, 2255–2264. [Google Scholar]
- Bartoli, G.; Cupone, G.; Dal Pozzo, R.; de Nino, A.; Mariuolo, L.; Procopio, A.; Tagarelli, A. Stereoselective reduction of enaminones to syn-γ-aminoalcohols. Tetrahedron Lett. 2002, 43, 7441–7444. [Google Scholar]
- Machado, M.A.; Harris, M.I.N.C.; Braga, A.C.H. Studies on the reduction of β-enamino ketones. J. Braz. Chem. 2006, 17, 1440–1442. [Google Scholar] [CrossRef]
- Cimarelli, C.; Giuli, S.; Palmieri, G. Stereoselective synthesis of enantiopure γ-aminoalcohols by reduction of chiral β-enaminoketones. Tetrahedron: Asymmetry 2006, 17, 1308–1317. [Google Scholar] [CrossRef]
- Cimarelli, C.; Palmieri, G.; Volpini, E. Regio- and stereoselective double alkylation of β-enamino esters with organolitium reagents followed by one-pot reduction: Convenient method for the synthesis of tertiary γ-amino alcohols. Tetrahedron 2006, 62, 9423–9432. [Google Scholar] [CrossRef]
- Elassar, A.-Z.A.; El-Khair, A.A. Recent developments in the chemistry of enaminones. Tetrahedron 2003, 59, 8463–8480. [Google Scholar] [CrossRef]
- Bartoli, G.; Cimarelli, C.; Marcantoni, E.; Palmieri, G.; Petrini, M. Chemo- and diastereoselective reduction of beta-enamino esters: A convenient synthesis of both cis- and trans-γ-amino alcohols and β-amino esters. J. Org. Chem. 1994, 59, 5328–5335. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Balbás, I.M.; Mendoza, B.E.D.; Fernández-Zertuche, M.; Ordoñez, M.; Linzaga-Elizalde, I. Synthesis of cis- and trans-3-Aminocyclohexanols by Reduction of β-Enaminoketones. Molecules 2012, 17, 151-162. https://doi.org/10.3390/molecules17010151
Balbás IM, Mendoza BED, Fernández-Zertuche M, Ordoñez M, Linzaga-Elizalde I. Synthesis of cis- and trans-3-Aminocyclohexanols by Reduction of β-Enaminoketones. Molecules. 2012; 17(1):151-162. https://doi.org/10.3390/molecules17010151
Chicago/Turabian StyleBalbás, Iris Montoya, Blanca Eda Domínguez Mendoza, Mario Fernández-Zertuche, Mario Ordoñez, and Irma Linzaga-Elizalde. 2012. "Synthesis of cis- and trans-3-Aminocyclohexanols by Reduction of β-Enaminoketones" Molecules 17, no. 1: 151-162. https://doi.org/10.3390/molecules17010151
APA StyleBalbás, I. M., Mendoza, B. E. D., Fernández-Zertuche, M., Ordoñez, M., & Linzaga-Elizalde, I. (2012). Synthesis of cis- and trans-3-Aminocyclohexanols by Reduction of β-Enaminoketones. Molecules, 17(1), 151-162. https://doi.org/10.3390/molecules17010151