1. Introduction
In current molecular biology and biotechnology, vectors are constructed by digesting plasmid DNA with restriction enzymes, followed by connection of this vector with predetermined gene fragment using ligase. In 1990, Aslanidis and de Jong proposed a system that may dramatically simplify this process [
1]. In this ligation-independent cloning (LIC) of PCR products, unnaturally long sticky ends of around 10–12-mer are formed on the ends of both a vector and a target gene fragment. Even if they are not ligated
in vitro, simple mixing of these fragments gives a quite stable vector-insert complex that can come into competent cells, and let the nicks be repaired by cells’ inherent repair pathways. The most important advantage of LIC is that it can eliminate any need to use restriction enzymes, which basically allows any gene to be cloned regardless of its sequence. To date, several methods to provide such unnaturally long sticky-ends to PCR products have been reported [
1,
2,
3,
4,
5,
6]. However, most of these techniques still require special design of sticky ends, additional time and budget-consuming enzymatic processing of PCR products, which often limit the scope of applications of LIC.
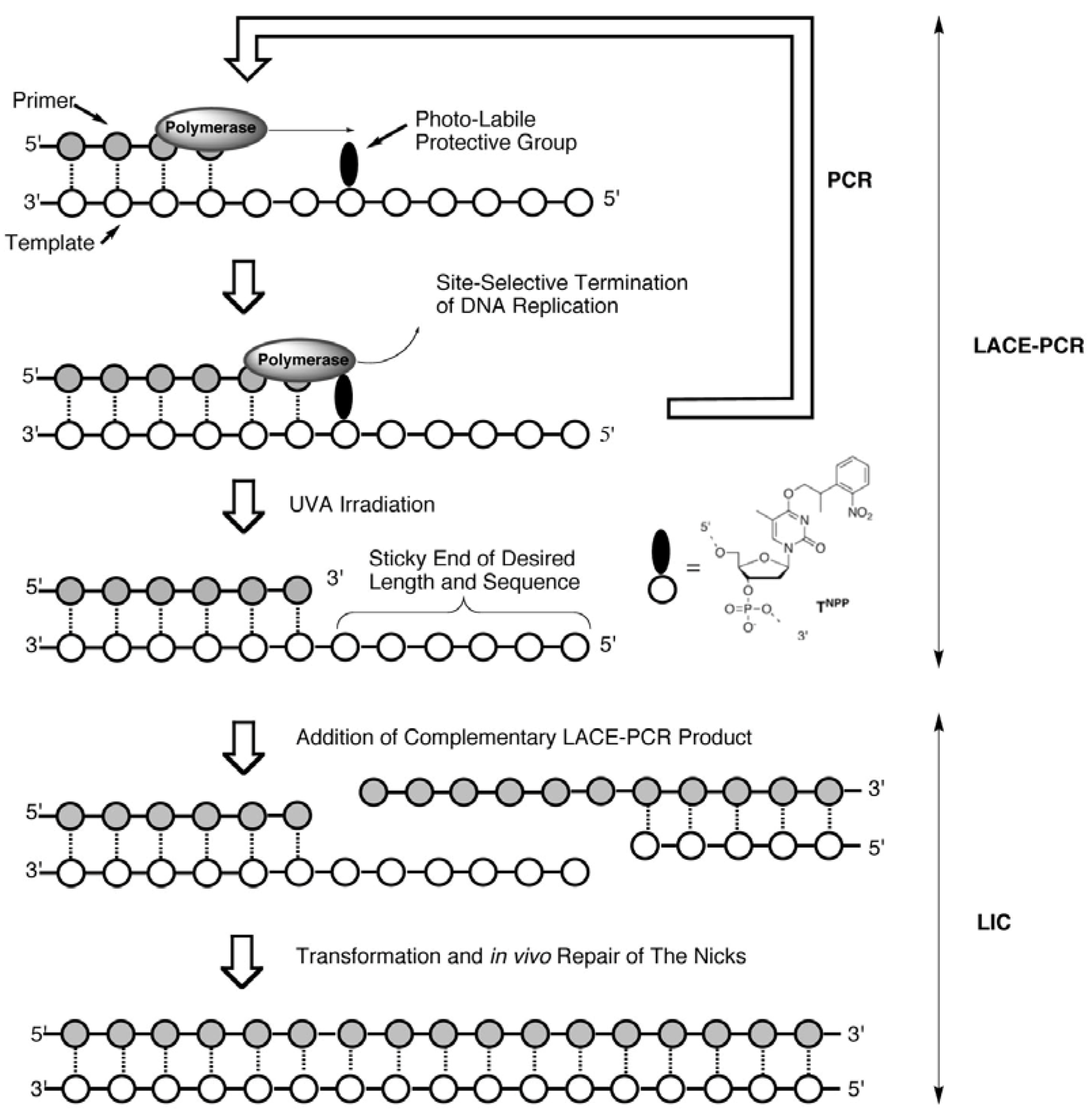
Recently, we have developed a quite simple and effective new chemistry-based PCR system, Light-Assisted Cohesive-Ending (LACE) PCR, that directly gives product with any desired sticky ends on both its ends (
Scheme 1) [
7,
8,
9]. In order to terminate a polymerase reaction at a desired position, a protected nucleotide with a photolabile protective group (a caged nucleotide) is incorporated into PCR primers [
10]. Within PCR cycles, elongation of the nascent strand (5′→3′ direction) is site-selectively terminated at the 3′-side of the caged nucleotide. Accordingly, the 5′-portion in the primer from the caged nucleotide remains single-stranded throughout the cycles, and predetermined sticky ends are obtained after the removal of the protecting group by brief UVA irradiation. No enzyme treatment on PCR product is necessary for the preparation of sticky-ends in this system.
Scheme 1.
A schematic illustration of LACE-PCR and LIC.
Scheme 1.
A schematic illustration of LACE-PCR and LIC.
The usefulness of LACE-PCR has been shown first in a conventional cloning using ligase with a sticky-ended plasmid prepared with restriction enzymes and an insert prepared with LACE-PCR [
8,
9], and recently in introducing point mutation in a plasmid as a variation of QuickChange
TM technique [
11], which involves simple self-circularization of sticky-ended products [
12]. Potential difficulties have been expected, on the other hand, in the application of LACE-PCR to LIC. For example, incomplete photo-removal of the caging group inhibits nick repair in cells exponentially with the number of the fragments to be joined. Possibility of DNA damages induced by UVA also increases as longer LACE-PCR products are required for a vector fragment [
13,
14,
15,
16,
17,
18].
In this study, we have examined various types of recombination including a construction of a gene encoding green fluorescent protein (GFP)-blue fluorescent protein (BFP) fusion protein, and confirmed that LIC is successfully achievable with LACE-PCR products, despite the above potential difficulties.
2. Results and Discussion
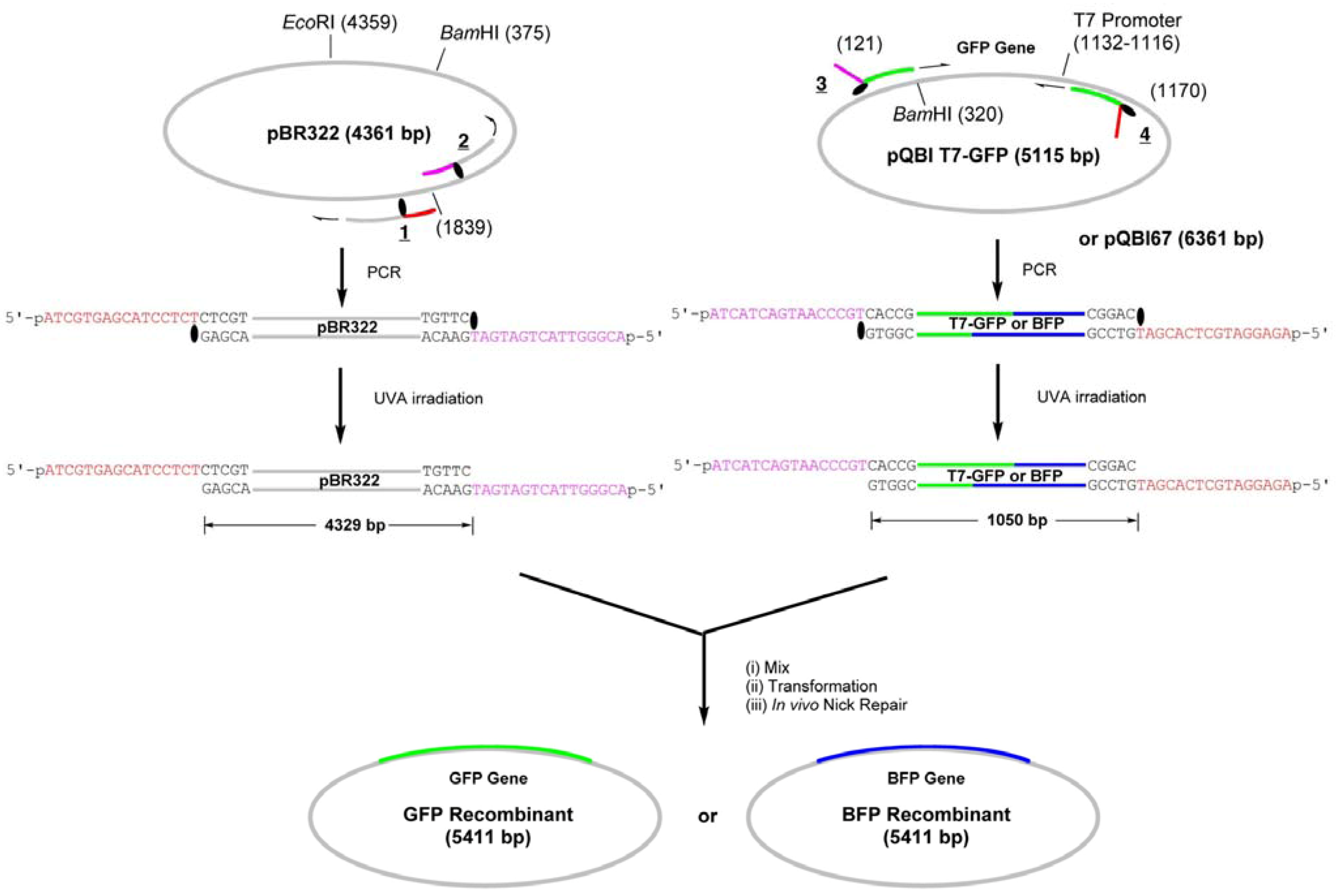
We have first constructed simple GFP-recombinant vector by amplifying the whole strand of pBR322 plasmid by LACE-PCR and inserting GFP gene, which is amplified from pQBI-T7GFP vector, into it (
Scheme 2).
Scheme 2.
Construction of GFP- or BFP-recombinant by LIC of LACE-PCR products.
Scheme 2.
Construction of GFP- or BFP-recombinant by LIC of LACE-PCR products.
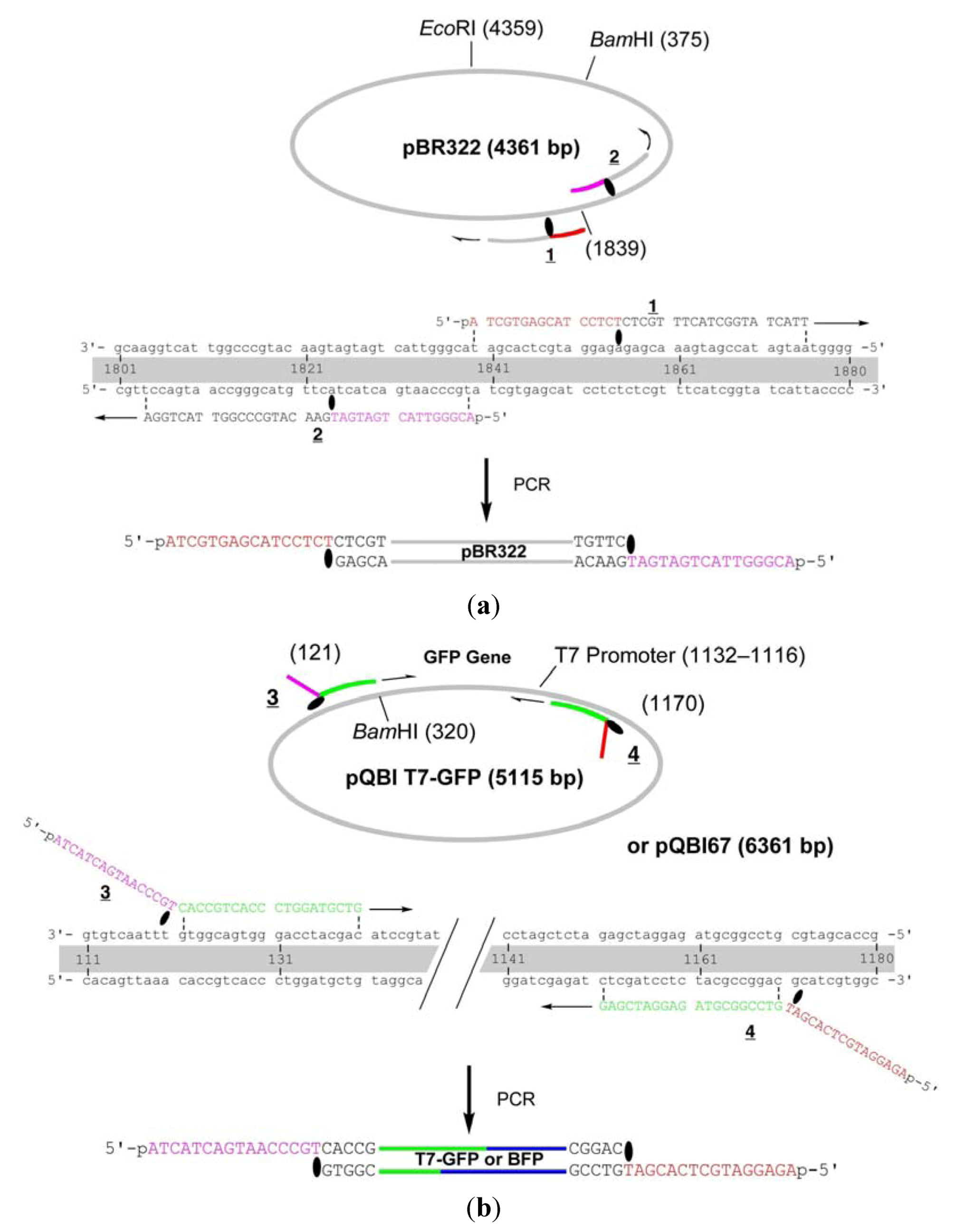
The primers
1 and
2 are complementary to pBR322 and used to amplify the whole strand of the vector (
Figure 1). These primers were chemically 5′-phosphorylated for the purpose of aiding repair in the cells, although it turned out in the construction of the fusion recombinant that the 5′-phosphates are not essential in the process (see below). The primer
1 hybridizes to 1840–1875-bp region and a caged thymidine, 4-
O-(2-(2-nitrophenyl)propyl)thymidine developed by Heckel
et al. (T
NPP in
Scheme 2) [
19] is incorporated to the position of T1855. This primer is completely complementary (except for the NPP introduction) to the plasmid, but the polymerase reaction in PCR is site-selectively blocked by the T
NPP [
7].
Figure 1.
Detailed structure of the primers for the construction of GFP- and BFP-recombinants. (a) Primers for the vector fragment;(b) Primers for the insert.
Figure 1.
Detailed structure of the primers for the construction of GFP- and BFP-recombinants. (a) Primers for the vector fragment;(b) Primers for the insert.
Thus, the complementary part of the 15 nt in the 5′ side of TNPP is not synthesized, and this portion serves as 16-nt sticky end after LACE-PCR. The primer 2, which has TNPP as the 16th nucleotide, binds to 1804–1839-bp region and similarly produces 16-nt sticky end with the sequence of 1824–1839-bp region. GFP gene was amplified by using 3 and 4, which are designed to hybridize to 121–140-bp and 1151–1170-bp regions in pQBI T7-GFP. The product obtained with these primers contains T7 promoter, open reading frame (ORF), and T7 terminator. To the 5′ end of 3, 16-nt stretch containing TNPP and complementary to the sticky end in 2 is attached. Similarly, 16 nt complementary to the sticky end in 1 is attached to the 5′ end of 4.
PCR amplification of the fragments was performed by using
Pfu polymerase, one of the most popular high-fidelity polymerases. The product was purified with a commercially available kit as Tris-EDTA (TE) buffered solution (pH 8.5), and then irradiated with UVA (300 < λ < 400 nm) for 15 min to remove NPP. As shown in
Figure 2, fairly pure products of expected length were obtained with the above procedure for both of GFP- and the vector fragments.
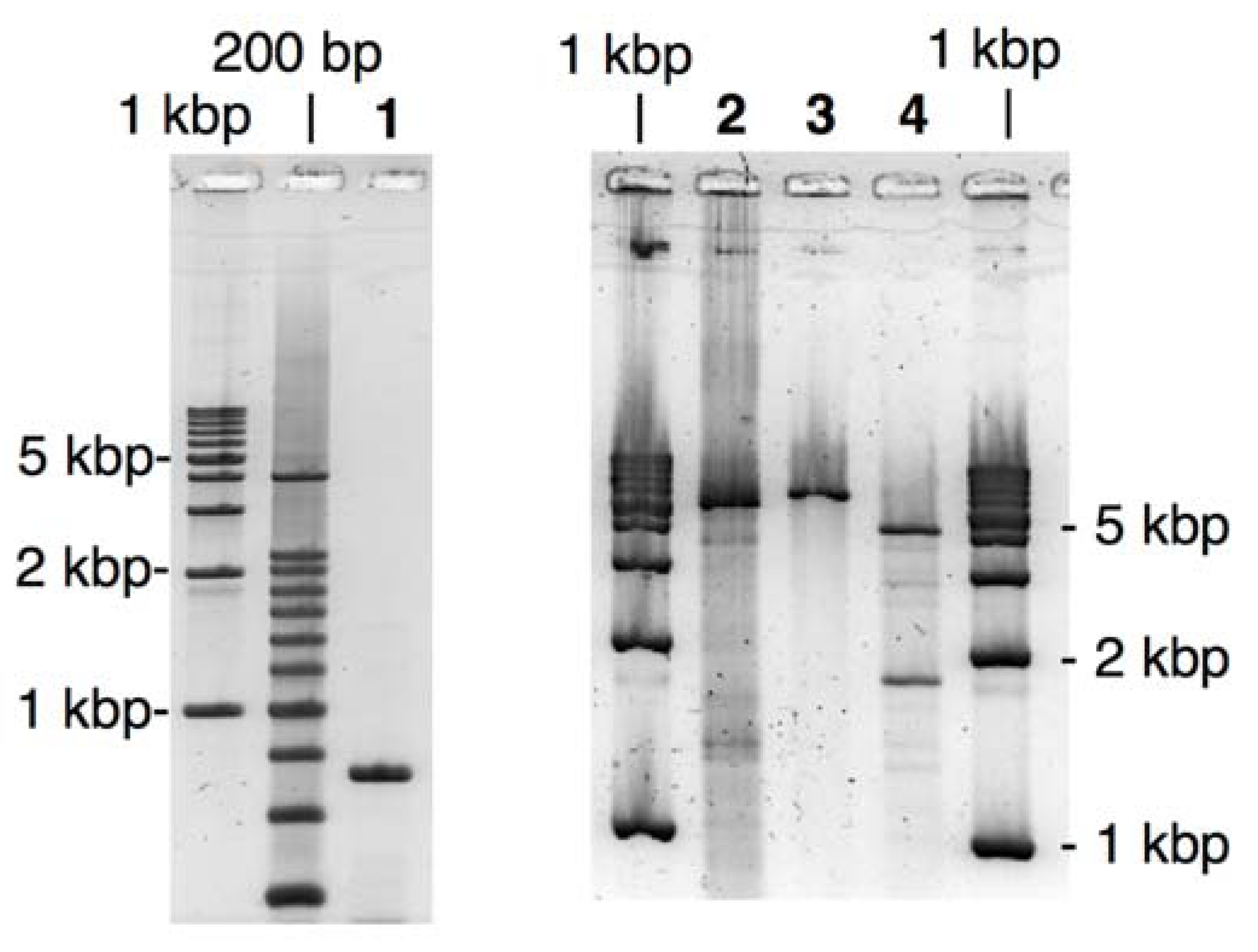
Figure 2.
Agarose-gel analysis of LACE-PCR products and the extracted recombinant plasmids for GFP- and BFP-recombinants. Lane 1, GFP fragment; Lane 2, BFP fragment; Lane 3, the vector fragment; Lane 4, GFP recombinant linearized with EcoRI; Lane 5, GFP recombinant treated with BamHI; Lane 6, BFP recombinant linearized with EcoRI; Lane 7, BFP recombinant treated with BamHI.
Figure 2.
Agarose-gel analysis of LACE-PCR products and the extracted recombinant plasmids for GFP- and BFP-recombinants. Lane 1, GFP fragment; Lane 2, BFP fragment; Lane 3, the vector fragment; Lane 4, GFP recombinant linearized with EcoRI; Lane 5, GFP recombinant treated with BamHI; Lane 6, BFP recombinant linearized with EcoRI; Lane 7, BFP recombinant treated with BamHI.
Transformation of E. coli using these LACE-PCR products was performed without any ligation process as follows: the GFP fragment (2 molar eq. ratio) was simply added to the solution of the vector fragment and incubated for 1 h at 37 °C. This mixture was then used for standard transformation of JM109 competent cells. Note that no enzymatic or chemical treatment was performed here. After overnight incubation at 37 °C, more than 100 colonies were obtained on an agar plate containing carbenicillin. Randomly selected 16 colonies were subjected to colony direct PCR using primers complementary to pBR322, 200 bp away from each of the joints, and eight of them gave the expected 1.4-kbp product from the desired GFP recombinant.
Construction of the recombinant coding BFP gene was performed in a same manner using the LACE-product from pQBI67. Within 16 colonies tested by direct PCR, 5 colonies were positive. Lanes 4–7 in
Figure 2 show an analysis of the extracted plasmid from the positive colonies. The vector fragment amplified by LACE-PCR contains one
EcoRI and one
BamHI cleavage site, and both of the GFP and BFP fragments contain one
BamHI site. If the complex of the vector and the insert fragment is successfully repaired in
E. coli and the desired recombinant vector is formed there, the extracted plasmid should give one linearized band of 5.4 kbp after
EcoRI digestion and two fragmented bands of 1.6 kbp and 3.8 kbp after
BamHI digestion. All of the observed bands in
Figure 2 are consistent with this argument. Moreover, sequencing of the extracted 10 plasmids has clearly shown that the nick between the two fragments were properly repaired in
E. coli and no mutation or deletion was induced around the joints and in ORF (see
Supporting Information).
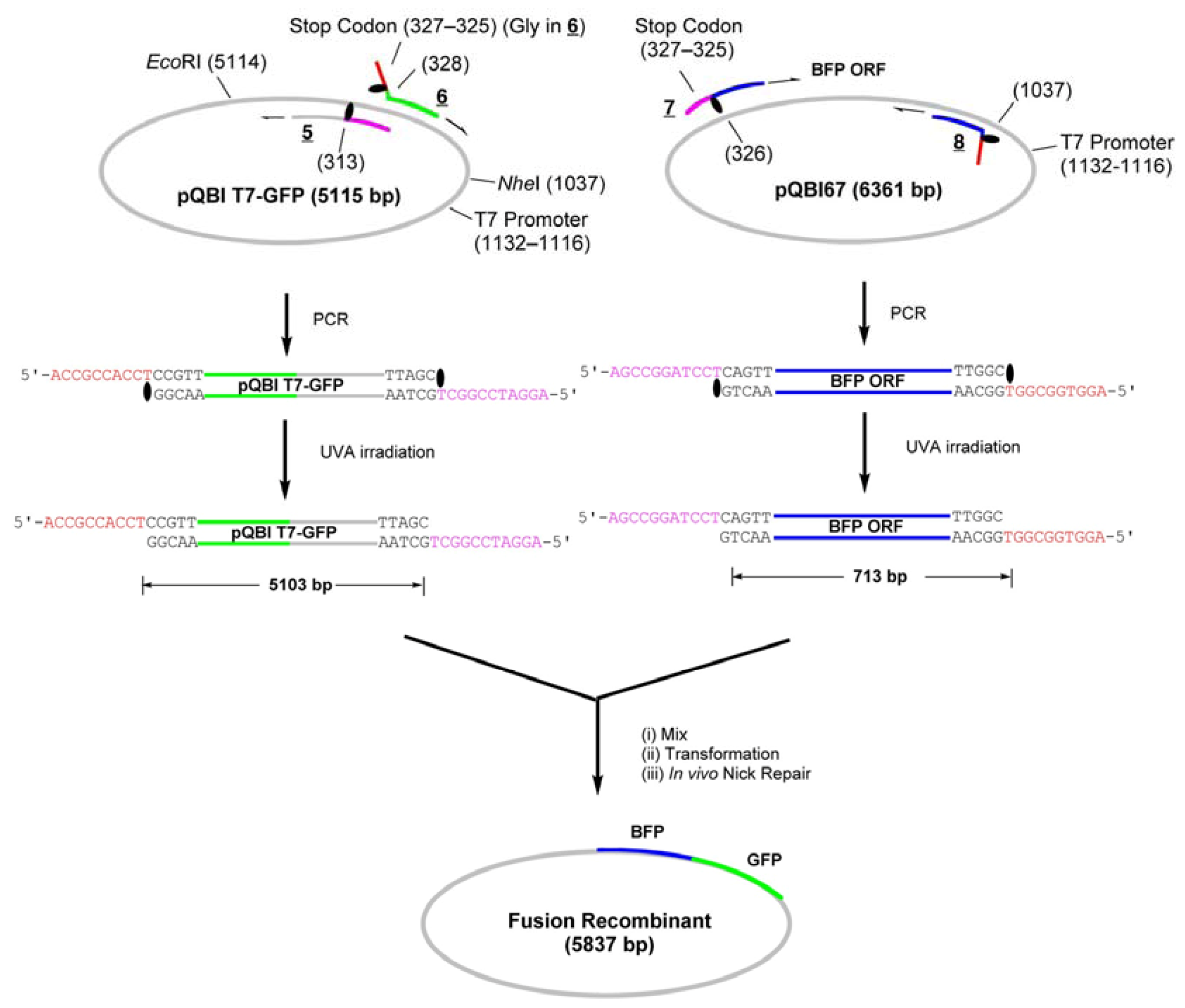
In addition to the above simple insertion of a gene to a vector, we have examined a possibility of making a fusion protein by using LACE-PCR without ligation. The homology between GFP and BFP is nearly 98%. They may thus be inappropriate substrates for the systems utilizing homologous recombination mechanisms such as In-Fusion technology [
6]. For this purpose, ORF of BFP was amplified from pQBI67 and inserted to the end of GFP gene in pQBI T7-GFP (
Scheme 3).
Scheme 3.
Construction of the fusion-recombinant encoding GFP-BFP fusion protein by LIC of LACE-PCR products.
Scheme 3.
Construction of the fusion-recombinant encoding GFP-BFP fusion protein by LIC of LACE-PCR products.
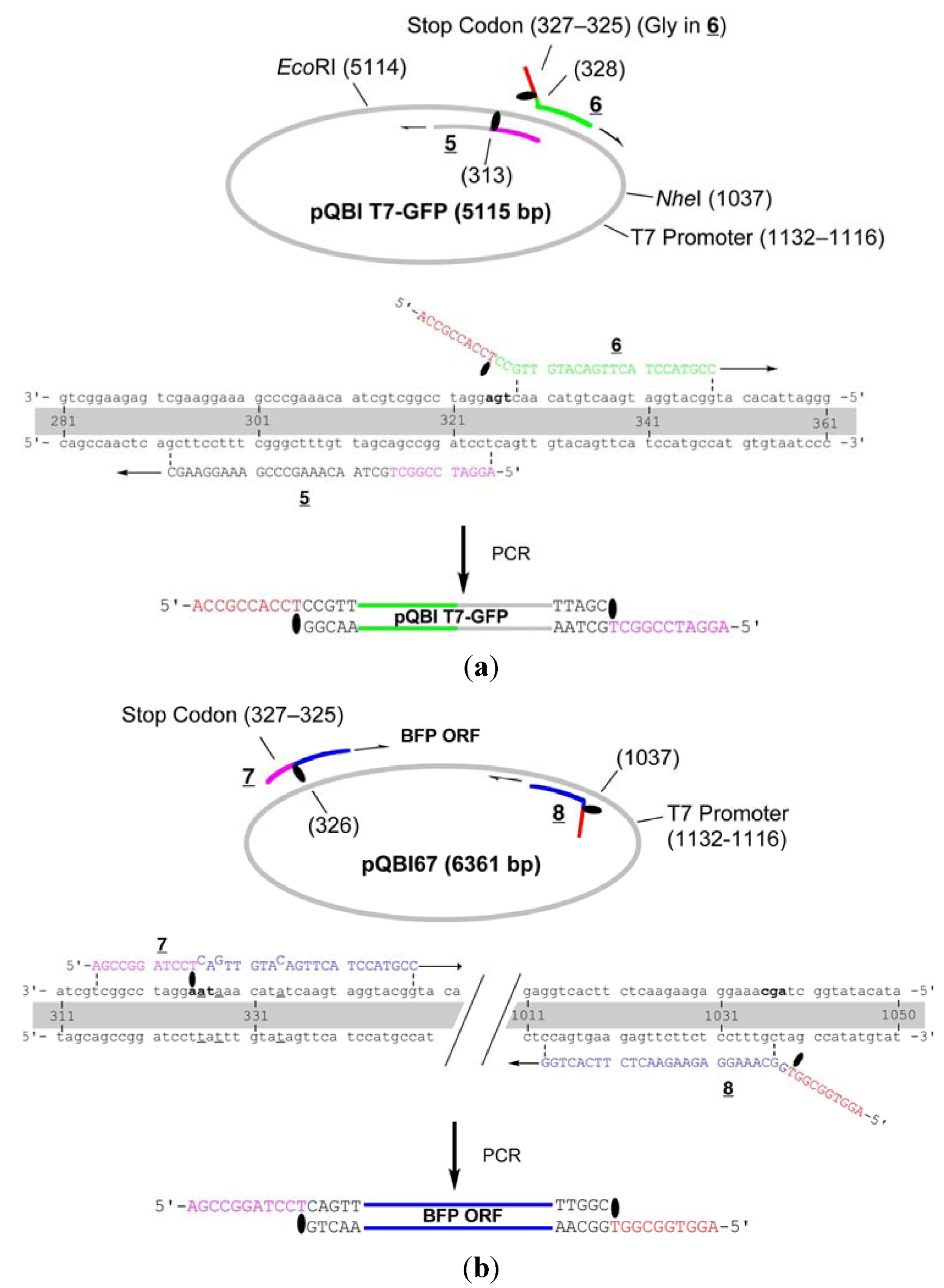
The whole strand of pQBI T7-GFP except for the 314–327-bp region, which contains an opal stop codon for GFP, were first amplified by LACE-PCR. The caged primers used in this experiment were not phosphorylated. The primers
5 and
6, which were used to amplify the vector, hybridize adjacent to this omitted region (
Figure 3). The primer 6 hybridizes to 291–313-bp region and has T
NPP in front of A314. In the 5′ side of T
NPP, 10-nt stretch with the sequence of 315–325-bp region is added. The primer 5 hybridizes to 328–348-bp region. In this primer, the nucleotide corresponding to A327 in the plasmid is changed to dC in order to mutate the opal stop codon to Gly. In addition, 9-nt stretch that corresponds to Gly ×3 and T
NPP is attached to the 5′ end. This portion serves as the sticky end after LACE-PCR, and as a part of Gly ×5 linker between GFP and BFP. Since the only requirement in LACE-PCR is the introduction of single caged nucleotide in a primer, such alteration of amino-acid sequence around the ends of the fragment is quite easy.
Figure 3.
Detailed structure of the primers for the construction of the fusion recombinant. (a) Primers for the vector fragment; (b) Primers for the insert. The opal stop codon in pQBI T7-GFP, ochre in pQBI67, and the codon for the third amino residue in ORF of GFP (Ser) are shown in boldface. The alteration in the sequence of pQBI67 from that of pQBI T7-GFP (T326. T328, and T334) is underlined.
Figure 3.
Detailed structure of the primers for the construction of the fusion recombinant. (a) Primers for the vector fragment; (b) Primers for the insert. The opal stop codon in pQBI T7-GFP, ochre in pQBI67, and the codon for the third amino residue in ORF of GFP (Ser) are shown in boldface. The alteration in the sequence of pQBI67 from that of pQBI T7-GFP (T326. T328, and T334) is underlined.
The primers
7 and
8 are designed to amplify ORF (325–1035-bp region) of BFP coded on pQBI67. The primer
7 hybridizes to 326–348-bp region and has T
NPP in front of A325. In the 5′ side of T
NPP, 10-nt stretch with the sequence of 314–324-bp region is added. Because of the sequence difference between pQBI T7-GFP and pQBI67, T326, T328, and T334 in pQBI67 are mismatched with
7. With the aid of this 10-nt stretch, however,
7 binds to the template strong enough to give fair amount of the desired product as shown below. The primer
8 hybridizes to 1013–1037-bp region. To mutate the third amino residue in BFP (Ser) to Gly, dG is incorporated in front of T1038 in the plasmid, and 9-nt stretch that is complementary to the Gly ×3 linker attached to
5 is introduced to the 5′ end. The size of the products obtained after LACE-PCR using these primers were all consistent with the design (lanes 1 and 2 in
Figure 4).
Figure 4.
Agarose-gel analysis of LACE-PCR product and the extracted recombinant plasmid for the fusion recombinant. Lane 1, ORF fragment of BFP; Lane 2, the vector fragment; Lane 3, the fusion recombinant linearized with EcoRI; Lane 4, the fusion recombinant treated with EcoRI and NheI.
Figure 4.
Agarose-gel analysis of LACE-PCR product and the extracted recombinant plasmid for the fusion recombinant. Lane 1, ORF fragment of BFP; Lane 2, the vector fragment; Lane 3, the fusion recombinant linearized with EcoRI; Lane 4, the fusion recombinant treated with EcoRI and NheI.
Transformation of JM109 using the simple mixture of both of these fragments was performed in a similar manner as described above. In direct-PCR test, 80% of the colonies obtained after overnight incubation were positive. This improved yield is probably because of the extended
DpnI treatment and the resulting complete elimination of the template plasmid. As shown in
Figure 4, the extracted recombinant plasmid linearized with
EcoRI (lane 3) was significantly longer than the LACE-PCR product of pQBI T7-GFP (lane 2). When the recombinant plasmid was double digested with
EcoRI and
NheI, two bands with the expected mobility (1.9- and 4.0-kbp fragments) were clearly observed. The sequence around the joints and ORF again completely agreed with the design (see
Supporting Information).
The extracted plasmids obtained above were further used to transform BL21-Gold (DE3) competent cells, in which T7 polymerase is expressed.
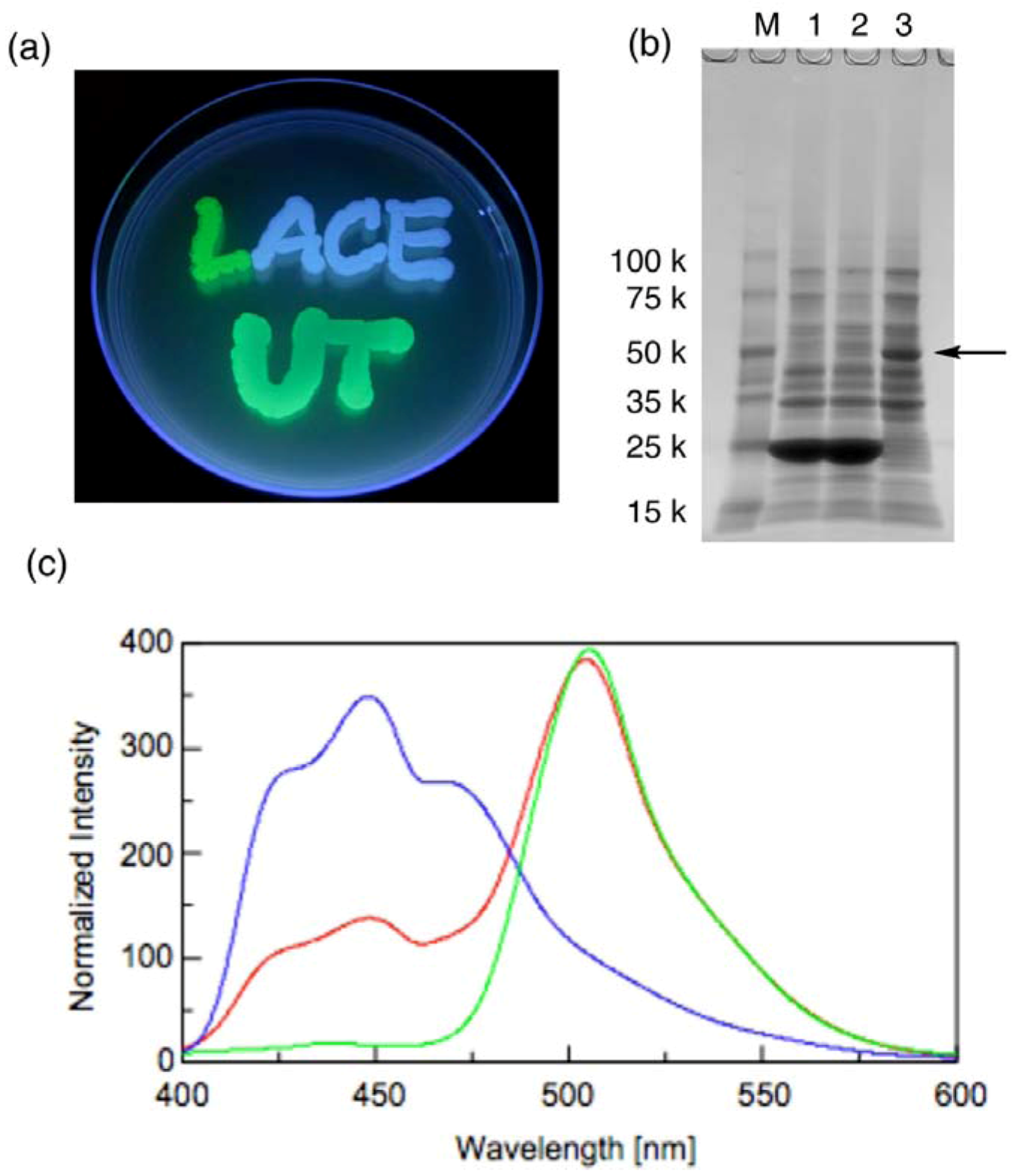
Figure 5a shows the emission of green fluorescence from the expressed GFP from the GFP-recombinant (the letter “L”), blue fluorescence from the expressed BFP from the BFP-recombinant (the letters “ACE”) and bluish green fluorescent from the expressed GFP-BFP fusion protein from the fusion recombinant (the letters “UT”) in each of the transformed cells. Successful fluorescence emission from each of the recombinants supports the sequencing analyses. The expressed proteins were further analyzed by preparing lysate of these transformed cells. The size of each protein was confirmed by SDS-PAGE of such lysate (
Figure 5b).
Figure 5.
Analyses of expressed fluorescent proteins. (a) Fluorescence from the three types of transformed E. coli; (b) SDS-PAGE of the lysate. Lane 1, Cells transformed with GFP-recombinant; Lane 2, BFP-recombinant; Lane 3, the fusion recombinant; M, size marker; (c) Normalized fluorescence emission spectra of the lysate. Green line, GFP-recombinant; blue line, BFP-recombinant; red line, the fusion recombinant. The excitation wavelength is 380 nm.
Figure 5.
Analyses of expressed fluorescent proteins. (a) Fluorescence from the three types of transformed E. coli; (b) SDS-PAGE of the lysate. Lane 1, Cells transformed with GFP-recombinant; Lane 2, BFP-recombinant; Lane 3, the fusion recombinant; M, size marker; (c) Normalized fluorescence emission spectra of the lysate. Green line, GFP-recombinant; blue line, BFP-recombinant; red line, the fusion recombinant. The excitation wavelength is 380 nm.
Very dark bands with mobility close to the 25-kDa marker, which is consistent with the expected molecular weight of native GFP and BFP (28 kDa), were observed for the lysate of GFP and BFP recombinants. For the lysate of the fusion recombinant, on the other hand, this band completely disappeared and a new significant band was observed near the 50-kDa marker. Fluorescence emission spectra of the lysate of the fusion recombinant showed two emission maxima at 449 nm and 504 nm, which correspond to the emission from BFP and GFP, respectively (
Figure 5c). The emission at 504 nm was significantly stronger than that at 449 nm. Considering that the excitation maximum wavelength of GFP is 473 nm, the large difference of the emission intensity may be attributable to fluorescence resonance energy transfer (FRET) between the two fluorophores, which is another evidence of successful construction of the fusion protein. All of these facts indicate that no critical mutation was induced in the regulatory elements such as promoter, ribosome-binding sites,
etc. in relatively longer vector fragments as well as ORF in the inserts.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







